
Abstract
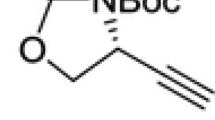
The ethynylglycine synthon {(R)-2,2-dimethyl-3-(tert-butoxycarbonyl)-4-ethynyl-oxazolidine} is a chiral compound with valuable synthetic interest. An update (covering literature from 2005 to 2017) on the different synthetic utilities is reviewed and discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and goals
In 2015, we decided to update the knowledge on ethynylglycine synthon 1a since our latest review in 2005 (Reginato et al. 2005b). The recently published part I of this review (Benfodda et al. 2015) dealt with the preparations of this compound (and its derivatives) from Garner’s aldehyde 2a described in the literature so far (Fig. 1). The synthetic strategy adopted, the optical purity, and the preferred protection for the amino protecting group have been discussed there. The present part II of this review will be devoted to the uses of ethynylglycine synthon in synthesis that have been reported since our 2005 review (2005–2017), showing the broad range of recent synthetic applications of this polyfunctional chiral synthon.

(R)-2,2-dimethyl-3-(tert-butoxycarbonyl)-4-ethynyl-oxazolidine {ethynylglycine synthon} 1a and Garner’s aldehyde 2a
The data that appeared after 2005 and that were not cited previously by us in our 2005 review (Reginato et al. 2005b) will be developed in this review; the previous references already cited in the 2005 review will only be cited in the paragraph headings, but the chemistry will not be developed. While writing this part II review, we noticed that a limited number of results earlier than 2005 were not developed in our 2005 review (Reginato et al. 2005b), that is why they now appear in the present review.
In neighbouring topics, it should be pointed out that Jirgensons recently published a review on the methods for the synthesis of α-ethynylglycines derivatives described since 1996 (Bolsakova and Jirgensons 2016), the precedent review being our older report on β,γ-alkynyl α-amino acids. (Meffre and Le Goffic 1996). We also published recently a comprehensive review on synthesis of α-quaternary α-ethynyl α-amino acids (Boibessot et al. 2016b).
Due to the presence of the oxazolidine ring (used especially as a synthetic precursor of α-amino acids) and of the terminal alkyne moiety, that allows several synthetic transformations, ethynylglycine synthon 1a is a useful building block for the synthesis of compounds of biological interest. The review will be organized considering the reaction type performed on the terminal alkyne (Fig. 1).
Metallation and reaction with electrophiles (Reginato et al. 1995, 1997; Meffre et al. 1996; Serrat et al. 1999; Cabarrocas et al. 2000a, b, 2001; Dondoni et al. 2001)
Electrophile is an aldehyde
Historically, this reaction was the first to be reported in the field of ethynylglycine synthon chemistry, in 1990. Because the Corey–Fuchs strategy (Benfodda et al. 2015) from aldehyde ent-2a yields to ethynylglycine synthon lithium acetylide (Li+)-ent-1a, the latter was directly trapped with paraformaldehyde to give a propargyl alcohol derivative 3 (Chung and Wasicak 1990). This compound was further functionalized to lead to an oxotremorine analogue 4. Oxotremorine 5 is the active metabolite of tremorine, a muscarinic receptor agonist (Fig. 2).

Synthesis of the oxotremorine analogue 4
Compound 3 was also used in 2010 to obtain the allenic oxazolidine 7 via a copper-mediated ortho-(diphenylphosphanyl)benzoate (oDPPB)-directed SN2′ reaction with an excellent regioselectivity (SN2′/SN2 = 95/5) (Fig. 3) (Spangenberg et al. 2010).

Synthesis of the allenic oxazolidine 7 via SN2′ reaction
The lithium acetylide derived from 1a was also reacted to TBDMS protected salicylaldehyde to give alcohol 8 (no yield given) which was then subjected to a one-pot acid-catalyzed nucleophilic substitution, fluoride TBDMS desilylation and exo-dig cycloisomerization to 2,3-disubstituted benzofuran 10 (Raji Reddy et al. 2012) (Fig. 4). It is worth noting that B(C6F5)3 was used as acid catalyst instead of BF3.Et2O because of the presence of acid labile acetonide group in 8.

Synthesis of the 2,3-disubstituted benzofuran 10
The lithium acetylide derived from ent-1a was again used and converted to an alkynyl zinc reagent in the presence of excess zinc chloride, necessary to ensure a highly selective addition to aldehyde 11 in favor of the formation of the 1,2-syn alkoxide 12, followed by a ring opening of the epoxide activated by excess Lewis acid zinc chloride. α-C-(ethynylglycine)-galactoside 13 was thus obtained (Guillarme et al. 2006) (Fig. 5).

Synthesis of the α-C-(ethynylglycine)-galactoside 13
Electrophile is a carboxylate or an isocyanate
The lithium acetylide derived from ent-1a was also condensed with methylchloroformate (inverse addition method to avoid the formation of enyne due to deprotonation and acetone elimination) to obtain alkynoate ent-14. Hydrostannylation followed by Stille cross coupling with iodo tryptophan 16 and NBoc deprotection led to the synthesis of 17. Compound 17 is a precursor to (+)-asperazine, an alkaloid with cytotoxic activity against human leukemia (14 steps from ent-1a) (Govek and Overman 2007) (Fig. 6).

Synthesis of methyl enoate 17, precursor of (+)-asperazine
The alkynoate 14 (obtained by condensation of the lithium acetylide derived from 1a with methyl cyanoformate, Mander’s reagent) was also engaged in a cycloaddition with benzyl formhydroximate 18 to afford isoxazole 19 which is a precursor of isoxazole 20 (9 steps from 1a). Isoxazole 20 is the precursor of a tetracycline core structure (11 steps from 20) (Wzorek et al. 2012) (Fig. 7).

Synthesis of isoxazole 20, precursor of a tetracyclic core structure
The arylamide 22, also obtained by condensation of the lithium acetylide derived from 1a with isocyanate 21, was subjected to an In-mediated radical cyclization to yield oxindole 24, a possible precursor of TMC-95A, a naturally occurring proteasome inhibitor (Yanada et al. 2005) (Fig. 8). Indeed, looking at the configuration of the starting material 1a used by the authors, and at the configuration of the stereogenic carbon 8 in TMC-95A, it seems that using (R)-1a oxindole (R)-24 would be actually obtained, which is a precursor of a diastereoisomer of TMC-95A (Fig. 8).

Synthesis of oxindole 24, precursor of TMC-95A
Electrophile is an alkyl halide
Some time ago, our groups reported the synthesis of silylated amino acids using the ethynylglycine synthon (Meffre et al. 1996; Reginato et al. 1998, 1999). More recently, we described the synthesis of unsaturated amino acids containing an allyl silane moiety (Reginato et al. 2006), using a silylated alkyl halide as electrophile, the amino acid being obtained from the oxazolidine ring by oxidation (Fig. 9).

Synthesis of an allylsilane amino acid derivative 28
Finally, ethynylglycine synthon rac-1a was converted to cyclic carbamate 29. The terminus of the alkyne in 29 was functionalized with trimethyl silyl and phenyl groups to give 30 and 31 which were subjected to an allenic Alder-ene reaction to give unstable triene 34 and 35 (Brummond and Yan 2008) (Fig. 10).

Synthesis of conjugated trienes 34 and 35
Pd-catalyzed coupling reactions: Sonogashira couplings (Reginato et al. 1997; Crisp et al. 1997; Cameron and Khambay 1998)
The terminal alkyne moiety on ethynylglycine synthon 1a allows functionalization using the well-known Pd-catalyzed Sonogashira coupling reaction (Chinchilla and Nájera 2007).
Furanomycin 41 is an unusual amino acid containing a 2,5-dihydrofuran ring that presents antibiotic activity (Katagiri et al. 1967; von Nussbaum et al. 2006) (Fig. 11).

Structures of (+)-furanomycin 41 and tryprostatins A 42 and B 43
The 2,5-dihydrofuran ring in the furanomycin analogue 40 was synthesized using a gold-catalyzed cycloisomerization of α-hydroxyallene 38 as the key step to obtain the precursor bicyclic dihydrofuran 39. α-Hydroxyallene 38 is obtained from propargyl oxirane 37 by a copper-mediated SN2′-substitution. Propargyl oxirane 37 is prepared from the corresponding enyne 36 which is obtained in turn from ethynylglycine synthon 1a by a Sonogashira coupling with 1-bromocyclooctene (Fig. 12) (Erdsack and Krause 2007).

Synthesis of furanomycin analogue 40
Tryprostatins A (42) and B (43) are naturally occurring 2,3-disubstituted indoles that present antimitotic properties (Fig. 11) (Evidente et al. 2014).
Total synthesis of tryprostatin B (Fig. 13) starts with Sonogashira coupling of the aromatic iodide 45 on the terminal alkyne of ent-1a. After partial reduction and dehydration, ortho-alkenyl isocyanide 47 is obtained. The 2,3-disubstituted indole 49 is obtained from 47 in a one-pot process by a radical cyclisation using V70 (2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) as a radical initiator, followed by a Stille-type coupling reaction through the 2-stannyl indole intermediate 48. After protection/deprotection steps and oxidation, compound 51, key intermediate in the synthesis of tryprostatin B, is obtained. (Yamakawa et al. 2010, 2011, 2014). Tryprostatin A is obtained using the same strategy.

Synthesis of carboxylic acid 51, precursor of tryprostatine B 43
Tryptophan analogues are interesting compounds, because they are possible indoleamine 2,3-dioxygenase (IDO) inhibitors and could have application in the treatment of inflammatory and neurodegenerative diseases (Röhrig et al. 2010). N,O-protected d-Homotryptophan 58 and its sulfur analogue 57 have been synthesized by Sonogashira coupling between 3-iodoheteroarenes and ethynylglycine synthon 1a followed by reduction of the alkyne, oxidation of the alcohol, and esterification using diazomethane (Fig. 14) (Goswami et al. 2012b). The selenohomotryptophan 59 (Fig. 14) was synthesized by the same group using the same route (Goswami et al. 2013).

Synthesis of homotryptophane derivative 58
The same group described the synthesis of 2- and 3-indolylglycine derivatives and of their oxygen analogues using ethynylglycine synthon 1a as starting material. Indolylglycines are interesting scaffolds which are present in bis-indole alkaloids like dragmacidins and hamacanthins (Goswami et al. 2012a).
3-Indolylglycine 68 and its oxygen analogue derivative 69 (Fig. 15) were prepared starting from the internal silyl derivatives 60 and 61 in turn obtained from 1a using the literature procedure (Meffre et al. 1996). The key step is a Larock’s heteroannulation with 2-iodoaniline and 2-iodophenol derivatives, to obtain compounds 62 and 63. After desilylation and acid-catalyzed oxazolidine opening into N-Boc-protected amino alcohols 66 and 67, the N,O-protected 3-indolylglycine 68 and the oxygen analogue 69 are obtained by oxidation and esterification (Goswami et al. 2012a).

Synthesis of 3-indolylglycine 68 and oxygen analogue 69
Unfortunately, the synthesis of the 2-substituted indole derivatives using the same strategy failed, due to the incompatibility of Boc-protecting group. For example Larock’s iodocyclization conditions (I2, CH2Cl2) proved to be problematic because of Boc-protecting group participation (nucleophilic attack). In this case, compound 71 was formed instead of expected indole derivative 72 (Fig. 16) (Goswami et al. 2012a).

Nucleophilic attack of Boc group under Larock’s iodocyclisation condition: formation of cyclic carbamate 71 instead of indole 72
To solve this problem, ethynylglycine synthon 1a was transformed into ethynyloxazolidinone 73, through the removal of acetonide protection and reaction of the resulting amino alcohol with thionyl chloride (Fig. 17). Coupling of cyclic carbamate 73 under Sonogashira conditions gave compounds 74 and 75, which after Boc protection and carbamate opening gave N-Boc-protected amino alcohols 76 and 77, from which the N,O-protected 2-indolylglycine 78 and the oxygen analogue 79 are obtained by oxidation and esterification (Goswami et al. 2012a).

Synthesis of 2-indolylglycine derivative 78 and oxygen analogue 79
During the synthesis of the chronic obstructive pulmonary disease (COPD) biomarker (+)-desmosine 86, a cross-linking amino acid of elastin, two of the four amino acid moieties present in the structure are introduced simultaneously using a Sonogashira cross-coupling reaction on ethynylglycine synthon ent-1a, as a first step. Another Sonogashira coupling reaction using propargylglycine derivatives 81 introduces a third amino acid moiety. Hydrogenation and usual functional group transformations led to the pyridine derivative 83, which is alkylated with the ω-iodobutylglycine derivative 84 to give N-Boc-protected desmosine 85. (+)-desmosine 86 is obtained after deprotection (Fig. 18) (Usuki et al. 2012; Yamada et al. 2015).

Synthesis of (+)-desmosine 86
Compound 88 was prepared by Sonogashira coupling between the ethynylglycine synthon ent-1a and triflate 87 and used to prepare 8-hydroxy-3-substituted isocoumarine 89 using a gold(I)-catalyzed cyclization (Fig. 19) (Mallampudi et al. 2017).

Synthesis of isocoumarine derivative 89
Cycloaddition reactions on the terminal alkyne (Falorni et al. 1998; Giacomelli et al. 2003)
The copper(I)-mediated reaction between nitrones and terminal alkynes (Kinugasa reaction) is a well-known method used for the β-lactam ring formation (Comas-Barceló and Harrity 2017). This reaction when applied to ethynylglycine synthon 1a leads to the formation of lactam 91 only in traces, although the same reaction conducted on D-glyceraldehyde alkyne analogue 92 led to lactam 93 in 46% yield and with a good diastereoselectivity (Fig. 20) (Stecko et al. 2009).

Synthesis of β-lactams 91 and 93
The [3 + 2]-cycloaddition of alkynes with in situ generated difluoromethyl nitrile oxide 94 (obtained from oxime 95) leads to CF2H-isoxazoles. This reaction when applied to ethynylglycine synthon ent-1a lets to isoxazole 96, a precursor of the fluorinated isoxazole amino acid 97 (Fig. 21) (Khutorianskyi et al. 2017). Due to bioisosterism of CHF2 and OH groups, compound 97 is an analogue of ibotenic acid 98, a naturally occurring non-selective glutamate receptor agonist. (Frydenvang et al. 2010).

Synthesis of isoxazole amino acid derivative 97
For another similar reaction, see (Falorni et al. 1998; Giacomelli et al. 2003), already cited in our previous review (Reginato et al. 2005b).
Huisgen 1,3-dipolar cycloaddition of an alkyne and an azide is a well-known access to 1,2,3-triazoles (Huisgen 1963; Totobenazara and Burke 2015). N-styryl triazole 101 was obtained from ethynylglycine synthon ent-1a by Huisgen cycloaddition with azido styrene 99 (generated in situ from cinnamic acid 100, CAN and NaN3) (Kavitha et al. 2011) (Fig. 22).

Synthesis of triazole amino acid rhizobitoxine analogue 104
Protected triazole amino acid 104 was also synthesized by us using the Huisgen cycloaddition of alkyne 102 and azido alanine 103 derived from l-serine. Protected aminoalcohol 102 was obtained by acid-catalyzed opening of the oxazolidine ring of ethynylglycine synthon ent-1a. Invertion of the deprotection/cycloaddition sequence led to lower yields (Boibessot et al. 2016a). Compound 104 is an analogue of rhizobitoxine 105, a plant growth regulator and inhibitor of PLP-dependent enzymes cystathionine β-lyase and ACC synthase (Fig. 22) (Owens et al. 1968; Xiong and Fuhrmann 1996; Yasuta et al. 1999; Sugawara et al. 2006).
Arylglycines are an interesting class of unusual amino acids, because this moiety is present in the structures of important biologically active natural products (Mazuela et al. 2017).
These types of compounds have been prepared by Dötz benzannulation between Fischer chromium carbene complexes 106 and the alkyne functionality of the ethynylglycine synthon 1a (Fig. 23) (Pulley et al. 1999, 2005).

Synthesis of arylglycine derivatives 110
In the key benzannulation reaction, the use of ultrasounds was found to improve yields.
The last oxidation step of arylglycinols 109 to arylglycines 110 proved to be problematic: the best results were obtained with a Dess–Martin oxidation followed by sodium chlorite oxidation using the Cbz-protecting group. The same oxidation performed on one of the Boc-protected arylglycinols proceeded in a lower yield (see, note 21 in Pulley et al. 2005).
Compounds 111 and 112 are serotoninergic chroman-based ligands with good activity (Fig. 24) (Holmberg et al. 2004, 2005). Badarau et al. synthesized compound 117, the 3-amino-7-azabenzofuran analogue of 111 and 112, starting from racemic ethynylglycine synthon 1a (Badarau et al. 2009). After ring opening, substitution of the methyl sulfonate in triazine 114 gave 115 which, through an intramolecular hetero-Diels–Alder reaction between the triazine residue and the alkyne moiety, led to compound 116. The reaction was carried out under microwave conditions using Cbz-protected alkyne 113. It is worth noting that, due to the presence of a good leaving group, the same reaction led to the formation of an oxazolidinone when performed on Boc-protected amino alcool 102 (Fig. 22) (Badarau et al. 2009).

Synthesis of a 3-amino-7-azabenzofuran derivative 117 analogue of serotoninergic ligands 111 and 112
Finally, the cyclopropene glutamate analogue 120 was synthesized by an Rh-catalyzed cyclopropanation of ethyl diazoacetate and ethynylglycine synthon 1a as a key step. Deprotection of oxazolidine 118 and oxidation of the alcohol finally furnished amino ester 120 although in low yield (Fig. 25) (Kumar et al. 2016). Ester deprotection proved to be unsuccessful. The unstability of these derivatives is due to the presence of the cyclopropene moiety and the acidic α-proton which lead to the formation of the corresponding allene (see supporting information in the reference of the work).

Synthesis of a cyclopropene amino acid glutamate analogue derivative 120
Addition of mixed tributylstannyl cuprate to the terminal alkyne and Stille coupling reactions (Reginato et al. 1997, 2000; Crisp et al. 1997)
Addition of stannylcuprate 121 onto ethynylglycine synthon 1a gave the vinyl copper intermediate 122. Hydrolytic work-up led to the γ-stannylated (E)-ethenyloxazolidine 123 in very good yield (Reginato et al. 1997).
Stille coupling with vinyl bromide gave diene 124. The use of palladium acetate Pd(OAc)2 and triphenylarsine AsPh3 as ligand proved to be necessary to obtain diene 130 when 2-bromopropene is used.
Trapping the intermediate vinyl copper 122 with electrophiles gave β-substituted stannyl allylamines 125, 126, and 127. Again Stille couplings led to diene 128 and triene 129 (Reginato et al. 2005a) (Fig. 26).

Synthesis of diene and triene amino acid precursors 124, 128, 129, and 130
It is possible to obtain selectively the β-stannylated 131, the regioisomer of 123 (131/123 = 9:1), the two compounds being separated by chromatography, using hydrostannation of ethynylglycine synthon 1a (Fig. 27) (Lin and Kazmaier 2007).

Synthesis of functionalized amino alcohols and heterocycles by hydrostannation
Compound 131 was subjected to Stille coupling reactions to afford alkenes 132 and 133. Vinylketones 132 were used for Michael additions, while alkenes 133 led to protected aminoalcohols 135 after oxazolidine cleavage (in two steps).
This strategy was also used in the same paper to obtain chiral amino heterocycles by hydrostannation and ring-closing metathesis (RCM) (Fig. 28) (Lin and Kazmaier 2007).

Synthesis of functionalized amino alcohols and heterocycles by hydrostannation
For this purpose, dienes 137 and 138 were needed. Because acidic cleavage of the oxazolidine ring could not be performed on the stannylated oxazolidine 131 (Fig. 27), alkyne ent-102 was first obtained by acidic deprotection and hydrostannation was performed to give vinyl stannane 136 which was converted to dienes 137 and 138.
Stille coupling and ring-closing metathesis led to heterocycles 141–142 and 147–148 (Fig. 28).
Metal-catalyzed C–C bond-forming reaction to the terminal alkyne (other than Pd-catalyzed couplings)
Enantiomerically pure N-Boc-protected (R,R)-diaminosuberic acid 152 was synthesized using a copper-catalyzed dimerization of ethynylglycine synthon 1a as key step, followed by usual transformations (hydrogenation, deprotection, and oxidation). This strategy would allow a facile introduction of tritium or deuterium (Fig. 29) (Callahan et al. 2000).

Synthesis of protected diaminosuberic acid 152
It is possible to synthesize a C-glycosyl derivative using an indium-mediated alkynylation reaction between a glycal or sugar derivative and alkynyl iodides under Barbier conditions (Ayed et al. 2010a). This reaction, when applied to alkynyliodide 153 (derived from ethynylglycine synthon 1a) and carbonyl compound 154 (derived from gluconolactone), leads to propargylic alcohol 155 in 66% yield, as a mixture of diastereoisomers (Fig. 30) (Ayed et al. 2010b).

Synthesis of C-glycosylated derivatives and the amino protecting group issue
Unfortunately, when the same alkynyliodide 153 was treated with tri-O-acetyl-D-glucal 156, no coupling product was detected in the same reaction conditions. Instead of the C-glycosylated derivative 157, the cyclic compound 158 was formed in 91% yield because of Boc intramolecular cyclization (Fig. 30) (Ayed et al. 2010a) (see also the discussion about the amino protecting group issue in our precedent Part I report) (Benfodda et al. 2015).
Indeed, when different protecting groups in alkynyliodide 153 were used (compounds 159 and 161), the coupling products 160 and 162 (Ferrier-type rearrangement) could be formed exclusively in the α-anomeric form (Fig. 31) (Ayed et al. 2010b, a).

Synthesis of C-glycosylated derivatives and the amino protecting group issue
Finally, propargyl hydroxylamine 164 was obtained in 75% yield by a C–C bond-forming reaction using a Lewis acid/metal amide hybrid-catalyzed reaction of ethynylglycine synthon 1a with nitrone 163 (Fig. 32) (Yamashita et al. 2014).

Synthesis of propargyl hydroxylamine derivative 164
Miscellaneous
Ethynylglycine synthon 1a was converted to allenic carbamate 165 using the Crabbé reaction (Fig. 33) (Crabbe et al. 1985; Alcaide et al. 2013).

Synthesis of oxazinone 166
The 6-methylene 1,3-oxazinan-2-one 166 has then been obtained using a gold-catalyzed oxycyclization of the allene 165 at room temperature (6-endo-dig oxyauration, kinetically controlled product). The same reaction conducted at high temperature led to a complex mixture (Fig. 33).
It is worth noting that the same reactions were performed (among other allenic carbamates) on the proline derivative 167. In that case, the thermodynamically favored 1,3-oxazine-2-one 170 was formed at high temperature (6-exo-dig oxyauration) (Fig. 34) (Alcaide et al. 2013).

Synthesis of oxazinones 169 and 170
Ethynylglycine synthon 1a was converted to protected propargylamine alcohol 171 which was subjected to intramolecular Mitsunobu reaction to lead to aziridine 172. Bromoallene 174 was then synthesized from aziridine 172 via an acid-mediated ring opening reaction and bromination through mesylate 173 (Fig. 35) (Ohno et al. 2002).

Synthesis of 2-ethynyl aziridine 172, mixture of enantiomers
Bromoallene 174 was used as a model in the course of the study of the intramolecular amination reaction of chiral bromoallenes into 2-ethynylaziridines in basic conditions (for example (R) and (S)-172 resulted from syn- and anti-SN2′ processes when bromoallene 174 was treated with LHMDS) (Fig. 35) (Ohno et al. 2002).
To circumvent the Wittig strategy to prepare alkene 175 from Garner’s aldehyde 2a (that proved to be unreliable to some authors) (Belanger et al. 2009), ethynylglycine synthon 1a was hydrogenated to alkene 175 using Lindlar catalyst. Cross-coupling metathesis using Grubbs’ catalyst II led to alkene 177. This compound is an intermediate in the synthesis of cyclic peptide 178 as a complexing agent of poly(vinyl alcohol) (PVA). This chemistry is outside the scope of this review and will not be detailed here (Fig. 36) (Belanger et al. 2009).

Synthesis of cyclic peptide 178
Conclusion
This review shows the great potentiality of ethynylglycine synthon as a polyfunctional chiral building block available for the synthesis of biologically relevant compounds, from “simple” to more “complex” structures. The terminal alkyne moiety has been exploited in metallation and reaction with a large variety of electrophile; in metal-catalyzed coupling reactions (Pd: Sonogashira, Cu, In); in cycloaddition reactions on nitrones, nitrile oxides, azide (Huisgen reaction), Fischer chromium carbene complexes, triazines (hetero-Diels–Alder), and ethyl diazo acetate (Rh-catalyzed cyclopropenation); in additions of stannylcuprates followed by Stille coupling reactions. Most of the times, the integrity of the chirality in ethynylglycine synthon was maintained in the final compounds or used as a chiral inducer.
The well-known reactivity of the terminal alkyne together with the presence of the stable chiral aminoalcohol substructure explain the increasing use of the ethynylglycine synthon in recent years (2005–2017: 35 reports) and suggests a great future.
Abbreviations
- Ac:
-
Acetyl
- ACC synthase:
-
1-Aminocyclopropane-1-carboxylate synthase
- All:
-
Allyl
- Bn:
-
Benzyl
- Boc:
-
tert-Butoxycarbonyl
- Bu or n-Bu:
-
n-Butyl
- CAN:
-
Cerium ammonium nitrate
- Cbz:
-
Benzyloxycarbonyl
- dba:
-
Dibenzylideneacetone
- DCC:
-
N,N′-Dicyclohexylcarbodiimide
- DIBAL-H:
-
Diisobutylalumino hydride
- DIPA:
-
Diisopropylamine
- DIPEA:
-
Diisopropylethylamine
- DMAP:
-
4-Dimethylaminopyridine
- DMF:
-
Dimethylformamide
- DMP:
-
Dess–Martin periodinane
- ent-x :
-
Enantiomer of compound x
- Et:
-
Ethyl
- HMDS:
-
Hexamethyldisilazane
- IDO:
-
Indoleamine 2,3-dioxygenase
- LHMDS:
-
Lithium bis(trimethylsilyl)amide
- MCPBA:
-
m-Chloroperoxybenzoic acid
- Me:
-
Methyl
- Mts:
-
2,4,6-Trimethylbenzenesulfonyl
- MW:
-
Microwave
- NCS:
-
N-Chlorosuccinimide
- oDPPBA:
-
2-(Diphenylphosphino)benzoic acid
- oDPPB:
-
2-(Diphenylphosphino)benzoate
- Ph:
-
Phenyl
- PLP:
-
Pyridoxal phosphate
- PTSA:
-
p-Toluenesulfonic acid
- RCM:
-
Ring-closing metathesis
- SEM:
-
2-(Trimethylsilyl)ethoxymethyl
- TBAF:
-
Tetrabutylammonium fluoride
- TBAI:
-
Tetrabutylammonium iodide
- TBDMS:
-
tert-butyldimethylsilyl
- TBDPS:
-
tert-butyldiphenylsilyl
- t-Bu:
-
tert-butyl
- TEMPO:
-
2,2,6,6-Tetramethylpiperidine 1-oxyl
- TES:
-
Triethylsilyl
- Tf:
-
Triflate
- TFA:
-
Trifluoroacetic acid
- THF:
-
Tetrahydrofuran
- TMEDA:
-
N,N,N′,N′-tetramethyl ethylenediamine
- TMS:
-
Trimethylsilyl
- Ts:
-
4-toluenesulfonyl
References
Alcaide B, Almendros P, Quirós MT, Fernández I (2013) Gold-catalyzed oxycyclization of allenic carbamates: expeditious synthesis of 1,3-oxazin-2-ones. Beilstein J Org Chem 9:818–826. https://doi.org/10.3762/bjoc.9.93
Ayed C, Palmier S, Lubin-Germain N et al (2010a) Indium-mediated alkynylation of sugars: synthesis of C-glycosyl compounds bearing a protected amino alcohol moiety. Carbohydr Res 345:2566–2570. https://doi.org/10.1016/j.carres.2010.07.033
Ayed C, Picard J, Lubin-Germain N et al (2010b) Synthesis of alkynes and alkynyl iodides bearing a protected amino alcohol moiety as functionalized amino acids precursors. Sci China Chem 53:1921–1926. https://doi.org/10.1007/s11426-010-4072-2
Badarau E, Suzenet F, Fînaru A-L, Guillaumet G (2009) Synthesis of 3-amino-8-azachromans and 3-amino-7-azabenzofurans via Inverse electron demand Diels–Alder reaction. Eur J Org Chem 2009:3619–3627. https://doi.org/10.1002/ejoc.200900191
Belanger D, Tong X, Soumare S et al (2009) Cyclic peptide-polymer complexes and their self-assembly. Chem Eur J. 15:4428–4436. https://doi.org/10.1002/chem.200802337 S4428/1-S4428/6
Benfodda Z, Bénimélis D, Reginato G, Meffre P (2015) Ethynylglycine synthon, a useful precursor for the synthesis of biologically active compounds: an update. Amino Acids 47:271–279. https://doi.org/10.1007/s00726-014-1902-0
Boibessot T, Bénimèlis D, Jean M et al (2016a) Synthesis of a novel rhizobitoxine-like triazole-containing amino acid. Synlett 27:2685–2688. https://doi.org/10.1055/s-0036-1588300
Boibessot T, Bénimélis D, Meffre P, Benfodda Z (2016b) Advances in the synthesis of α-quaternary α-ethynyl α-amino acids. Amino Acids 48:2081–2101. https://doi.org/10.1007/s00726-016-2276-2
Bolsakova J, Jirgensons A (2016) Synthesis of α-Ethynyl Glycines. Eur J Org Chem 2016:4591–4602. https://doi.org/10.1002/ejoc.201600253
Brummond KM, Yan B (2008) Rhodium(I)-catalyzed cycloisomerization reaction of yne-allenamides: an approach to cyclic enamides. Synlett 2008:2303–2308. https://doi.org/10.1055/s-2008-1078169
Cabarrocas G, Rafel S, Ventura M, Villalgordo J (2000a) A new approach toward the stereoselective synthesis of novel quinolyl glycines: synthesis of the enantiomerically pure Quinolyl-β-amino alcohol precursors. Synlett 2000:0595–0598. https://doi.org/10.1055/s-2000-6625
Cabarrocas G, Ventura M, Maestro M et al (2000b) Reaction between hydrazines and chiral α-acetylenic ketones: synthesis of novel enantiomerically pure pyrazolyl-β-amino alcohols†. Tetrahedron Asymmetry 11:2483–2493. https://doi.org/10.1016/S0957-4166(00)00204-4
Cabarrocas G, Ventura M, Maestro M et al (2001) Synthesis of novel optically pure quinolyl-β-amino alcohol derivatives from 2-amino thiophenol and chiral α-acetylenic ketones and their IBX-mediated oxidative cleavage to N-Boc quinolyl carboxamides. Tetrahedron Asymmetry 12:1851–1863. https://doi.org/10.1016/S0957-4166(01)00308-1
Callahan JF, Khatana SS, Bhatnagar PK (2000) Stereoselective synthesis of diaminosuberic acid via a chiral alkynyl oxazolidine. Synth Commun 30:1213–1219. https://doi.org/10.1080/00397910008087141
Cameron S, Khambay BPS (1998) Stereospecific synthesis of the amino acid, (S)-2-amino-(Z)-3,5-hexadienoic acid. Tetrahedron Lett 39:1987–1990. https://doi.org/10.1016/S0040-4039(98)00112-9
Chinchilla R, Nájera C (2007) The sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem Rev 107:874–922. https://doi.org/10.1021/cr050992x
Chung JYL, Wasicak JT (1990) Synthesis of chiral α-acetylenic cyclic amines from α-amino acids: applications to differentially constrained oxotremorine analogues as muscarinic agents. Tetrahedron Lett 31:3957–3960. https://doi.org/10.1016/S0040-4039(00)94471-X
Comas-Barceló J, Harrity JPA (2017) Metal acetylides in cycloaddition reactions. Synthesis 49:1168–1181. https://doi.org/10.1055/s-0036-1588922
Crabbe P, Schlemper Elmer O, Kay Fair et al (1985) Allene Synthesis by organo-metallic reactions. Isr J Chem 26:147–151. https://doi.org/10.1002/ijch.198500085
Crisp GT, Jiang Y-L, Pullman PJ, De Savi C (1997) Elaboration of the side-chain of amino acid derivatives by palladium catalysed couplings. Tetrahedron 53:17489–17500. https://doi.org/10.1016/S0040-4020(97)10197-1
Dondoni A, Mariotti G, Marra A, Massi A (2001) Expeditious synthesis of β-linked glycosyl serine methylene isosteres (β-C-gly ser) via ethynylation of sugar lactones. Synthesis. https://doi.org/10.1055/s-2001-18058
Erdsack J, Krause N (2007) Synthesis of furanomycin derivatives by gold-catalyzed cycloisomerization of α-hydroxyallenes. Synthesis 2007:3741–3750. https://doi.org/10.1055/s-2007-990860
Evidente A, Kornienko A, Cimmino A et al (2014) Fungal metabolites with anticancer activity. Nat Prod Rep 31:617–627. https://doi.org/10.1039/C3NP70078J
Falorni M, Giacomelli G, Spanu E (1998) Synthesis of new α-amino- acids containing the isoxazole moiety. Tetrahedron Lett 39:9241–9244. https://doi.org/10.1016/S0040-4039(98)02010-3
Frydenvang K, Pickering DS, Greenwood JR et al (2010) Biostructural and pharmacological studies of bicyclic analogues of the 3-isoxazolol glutamate receptor agonist ibotenic acid. J Med Chem 53:8354–8361. https://doi.org/10.1021/jm101218a
Giacomelli G, De Luca L, Porcheddu A (2003) A method for generating nitrile oxides from nitroalkanes: a microwave assisted route for isoxazoles. Tetrahedron 59:5437–5440. https://doi.org/10.1016/S0040-4020(03)00859-7
Goswami K, Duttagupta I, Sinha S (2012a) Synthesis of optically active 2- and 3- indolylglycine derivatives and their oxygen analogues. J Org Chem 77:7081–7085. https://doi.org/10.1021/jo300708h
Goswami K, Paul S, Bugde ST, Sinha S (2012b) Synthesis of optically active homotryptophan and its oxygen and sulfur analogues. Tetrahedron 68:280–286. https://doi.org/10.1016/j.tet.2011.10.055
Goswami K, Chakraborty A, Sinha S (2013) Synthesis of optically active selenium-containing isotryptophan, homoiso-tryptophan, and homotryptophan. Eur J Org Chem 2013:3645–3647. https://doi.org/10.1002/ejoc.201300352
Govek SP, Overman LE (2007) Total synthesis of (+)-asperazine. Tetrahedron 63:8499–8513. https://doi.org/10.1016/j.tet.2007.05.127
Guillarme S, Plé K, Haudrechy A (2006) Selective synthesis of α-C-(Alkynyl)-galactosides by an efficient tandem reaction. J Org Chem 71:1015–1017. https://doi.org/10.1021/jo0519817
Holmberg P, Sohn D, Leideborg R et al (2004) Novel 2-aminotetralin and 3-aminochroman derivatives as selective serotonin 5-HT7 receptor agonists and antagonists. J Med Chem 47:3927–3930. https://doi.org/10.1021/jm0498102
Holmberg P, Tedenborg L, Rosqvist S, Johansson AM (2005) Novel 3-aminochromans as potential pharmacological tools for the serotonin 5-HT7 receptor. Bioorg Med Chem Lett 15:747–750. https://doi.org/10.1016/j.bmcl.2004.11.013
Huisgen R (1963) 1,3-Dipolar Cycloadditions. Past and Future. Angew Chem Int Ed Engl 2:565–598. https://doi.org/10.1002/anie.196305651
Katagiri K, Tori K, Kimura Y et al (1967) A new antibiotic. furanomycin, an isoleucine antagonist. J Med Chem 10:1149–1154. https://doi.org/10.1021/jm00318a035
Kavitha M, Mahipal B, Mainkar PS, Chandrasekhar S (2011) Click reaction on in situ generated β-azidostyrenes from cinnamic acid using CAN–NaN3: synthesis of N-styryl triazoles. Tetrahedron Lett 52:1658–1662. https://doi.org/10.1016/j.tetlet.2011.01.129
Khutorianskyi A, Chalyk B, Borysko P et al (2017) Difluoromethyl nitrile oxide (CF2HCNO): a neglected chemical reagent. Eur J Org Chem 2017:3935–3940. https://doi.org/10.1002/ejoc.201700764
Kumar P, Shukhman D, Laughlin ST (2016) A photocaged, cyclopropene-containing analog of the amino acid neurotransmitter glutamate. Tetrahedron Lett 57:5750–5752. https://doi.org/10.1016/j.tetlet.2016.10.106
Lin H, Kazmaier U (2007) Regioselective mo-catalyzed hydrostannations as key steps in the synthesis of functionalized amino alcohols and heterocycles. Eur J Org Chem 2007:2839–2843. https://doi.org/10.1002/ejoc.200700126
Mallampudi NA, Reddy GS, Maity S, Mohapatra DK (2017) Gold(I)-Catalyzed Cyclization for the Synthesis of 8-Hydroxy-3—substituted Isocoumarins: total Synthesis of Exserolide F. Org Lett 19:2074–2077. https://doi.org/10.1021/acs.orglett.7b00673
Mazuela J, Antonsson T, Johansson MJ et al (2017) Direct Synthesis of N-Alkyl Arylglycines by Organocatalytic Asymmetric Transfer Hydrogenation of N-Alkyl Aryl Imino Esters. Org Lett. https://doi.org/10.1021/acs.orglett.7b02627
Meffre P, Le Goffic F (1996) β, γ-Alkynylα-amino acids: a synthetic challenge. Amino Acids 11:313–328. https://doi.org/10.1007/BF00807939
Meffre P, Gauzy L, Branquet E et al (1996) Synthesis of optically active β, γ-alkynylglycine derivatives. Tetrahedron 52:11215–11238. https://doi.org/10.1016/0040-4020(96)00630-8
Ohno H, Ando K, Hamaguchi H et al (2002) A highly cis-selective synthesis of 2-ethynylaziridines by intramolecular amination of chiral bromoallenes: improvement of stereoselectivity based on the computational investigation. J Am Chem Soc 124:15255–15266. https://doi.org/10.1021/ja0262277
Owens LD, Guggenheim S, Hilton JL (1968) Rhizobium-synthesized phytotoxin: an inhibitor of β-cystathionase in Salmonella typhimurium. Biochim Biophys Acta BBA Gen Subj 158:219–225. https://doi.org/10.1016/0304-4165(68)90134-7
Pulley SR, Sen S, Vorogushin A, Swanson E (1999) Diaryl ethers using fischer chromium carbene mediated benzannulation. Org Lett 1:1721–1723. https://doi.org/10.1021/ol990949u
Pulley SR, Czakó B, Brown GD (2005) Synthesis of arylglycines via the Dötz benzannulation reaction. Tetrahedron Lett 46:9039–9042. https://doi.org/10.1016/j.tetlet.2005.10.105
Raji Reddy C, Krishna G, Kavitha N et al (2012) Access to 2,3-disubstituted benzofurans through one-pot acid-catalyzed nucleophilic substitution/TBAF-mediated oxacycloisomerization. Eur J Org Chem 2012:5381–5388. https://doi.org/10.1002/ejoc.201200708
Reginato G, Mordini A, Degl’Innocenti A, Caracciolo M (1995) Stereoselective synthesis of (R)-(−)-2,2-dimethyl-3-t-butoxycarbonyl-4-ethynyl-oxazolidine: a chiral building block for the synthesis of a new class of substituted alkynes. Tetrahedron Lett 36:8275–8278. https://doi.org/10.1016/0040-4039(95)01725-w
Reginato G, Mordini A, Caracciolo M (1997) Synthetic Elaboration of the Side Chain of (R)-2,2-dimethyl-3-(tert-butoxycarbonyl)-4-ethynyloxazolidine: a new regio—and stereoselective strategy to δ-functionalized β-amino alcohols. J Org Chem 62:6187–6192. https://doi.org/10.1021/jo970619s
Reginato G, Mordini A, Valacchi M (1998) A stereoselective approach to the synthesis of γ-silylated amino acids. Tetrahedron Lett 39:9545–9548. https://doi.org/10.1016/S0040-4039(98)02120-0
Reginato G, Mordini A, Valacchi M, Grandini E (1999) Silylcupration of (R)-2,2-Dimethyl-3-(tert-butoxycarbonyl)-4-ethynyloxazolidine: a stereoselective approach to the synthesis of γ-silylated saturated and unsaturated α-amino acids. J Org Chem 64:9211–9216. https://doi.org/10.1021/jo991272r
Reginato G, Mordini A, Verrucci M et al (2000) A new approach to non racemic saturated and unsaturated 5-aminoalkyl methyl ketones. Tetrahedron Asymmetry 11:3759–3768. https://doi.org/10.1016/S0957-4166(00)00335-9
Reginato G, Gaggini F, Mordini A, Valacchi M (2005a) Stereoselective synthesis of dienylamines: from amino acids to E-alkene dipeptide isosters. Tetrahedron 61:6791–6800. https://doi.org/10.1016/j.tet.2005.04.068
Reginato G, Meffre P, Gaggini F (2005b) Ethynylglycine synthon from Garner’s aldehyde: a useful precursor for the synthesis of non-natural amino acids. Amino Acids 29:81–87. https://doi.org/10.1007/s00726-005-0184-y
Reginato G, Mordini A, Meffre P et al (2006) New unsaturated amino acids containing an allylsilane moiety on the lateral chain. Tetrahedron Asymmetry 17:922–926. https://doi.org/10.1016/j.tetasy.2006.02.017
Röhrig UF, Awad L, Grosdidier A et al (2010) Rational design of indoleamine 2,3-dioxygenase inhibitors. J Med Chem 53:1172–1189. https://doi.org/10.1021/jm9014718
Serrat X, Cabarrocas G, Rafel S et al (1999) A highly efficient and straightforward stereoselective synthesis of novel chiral α-acetylenic ketones. Tetrahedron Asymmetry 10:3417–3430. https://doi.org/10.1016/S0957-4166(99)00357-2
Spangenberg T, Schoenfelder A, Breit B, Mann A (2010) 1,2-Diastereoselective C–C bond-forming reactions for the synthesis of chiral β-branched α-amino acids. Eur J Org Chem 2010:6005–6018. https://doi.org/10.1002/ejoc.201000865
Stecko S, Mames A, Furman B, Chmielewski M (2009) Asymmetric kinugasa reaction of cyclic nitrones and nonracemic acetylenes. J Org Chem 74:3094–3100. https://doi.org/10.1021/jo900121x
Sugawara M, Okazaki S, Nukui N et al (2006) Rhizobitoxine modulates plant–microbe interactions by ethylene inhibition. Biotechnol Adv 24:382–388. https://doi.org/10.1016/j.biotechadv.2006.01.004
Totobenazara J, Burke AJ (2015) New click-chemistry methods for 1,2,3-triazoles synthesis: recent advances and applications. Tetrahedron Lett 56:2853–2859. https://doi.org/10.1016/j.tetlet.2015.03.136
Usuki T, Yamada H, Hayashi T et al (2012) Total synthesis of COPD biomarker desmosine that crosslinks elastin. Chem Commun 48:3233–3235. https://doi.org/10.1039/C2CC17958J
von Nussbaum F, Brands M, Hinzen B et al (2006) Antibacterial Natural Products in Medicinal Chemistry—Exodus or Revival? Angew Chem Int Ed 45:5072–5129. https://doi.org/10.1002/anie.200600350
Wzorek JS, Knöpfel TF, Sapountzis I, Evans DA (2012) A macrocyclic approach to tetracycline natural products. investigation of transannular alkylations and michael additions. Org Lett 14:5840–5843. https://doi.org/10.1021/ol302691j
Xiong K, Fuhrmann JJ (1996) Comparison of rhizobitoxine-induced inhibition of β-cystathionase from different bradyrhizobia and soybean genotypes. Plant Soil 186:53–61. https://doi.org/10.1007/BF00035055
Yamada H, Hayashi T, Usuki T (2015) Total synthesis of the COPD biomarker desmosine via stepwise sonogashira Cross-coupling reactions. Bull Chem Soc Jpn 88:673–683. https://doi.org/10.1246/bcsj.20140394
Yamakawa T, Ideue E, Shimokawa J, Fukuyama T (2010) Total synthesis of tryprostatins A and B. Angew Chem Int Ed 49:9262–9265. https://doi.org/10.1002/anie.201004963
Yamakawa T, Ideue E, Iwaki Y et al (2011) Total synthesis of tryprostatin A and B. Tetrahedron 67:6547–6560. https://doi.org/10.1016/j.tet.2011.05.112
Yamakawa T, Ideue E, Shimokawa J, Fukuyama T (2014) Corrigendum: total synthesis of tryprostatins A and B. Angew Chem Int Ed 53:8808–8808. https://doi.org/10.1002/anie.201401055
Yamashita Y, Saito Y, Imaizumi T, Kobayashi S (2014) A Lewis acid/metal amide hybrid as an efficient catalyst for carbon-carbon bond formation. Chem Sci 5:3958–3962. https://doi.org/10.1039/C4SC01332H
Yanada R, Obika S, Kobayashi Y et al (2005) Stereoselective synthesis of 3-alkylideneoxindoles using tandem indium-mediated carbometallation and palladium-catalyzed cross-coupling reactions. Adv Synth Catal 347:1632–1642. https://doi.org/10.1002/adsc.200505147
Yasuta T, Satoh S, Minamisawa K (1999) New assay for rhizobitoxine based on inhibition of 1-aminocyclopropane-1-carboxylate synthase. Appl Environ Microbiol 65:849–852
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Research subjects
This review is a compilation of the previous works performed by different authors. No animal or human was used or harmed in this work.
Informed consent
This manuscript is being submitted after consent was obtained from all authors, and all authors are aware of this manuscript submission.
Additional information
Handling Editor: J. D. Wade.
Rights and permissions
About this article

Cite this article
Benfodda, Z., Benimélis, D., Reginato, G. et al. Ethynylglycine synthon, a useful precursor for the synthesis of biologically active compounds: an update. Part II: synthetic uses of ethynylglycine synthon. Amino Acids 50, 1307–1328 (2018). https://doi.org/10.1007/s00726-018-2628-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-018-2628-1