
Abstract
To date, there have been few reports analyzing the amino acid requirement for growth of hyperthermophilic archaea. We here found that the hyperthermophilic archaeon Pyrococcus horikoshii OT-3 requires Thr, Leu, Val, Phe, Tyr, Trp, His and Arg in the medium for growth, and shows slow growth in medium lacking Met or Ile. This largely corresponds to the presence, or absence, of genes related to amino acid biosynthesis in its genome, though there are exceptions. The amino acid requirements were dramatically lost by addition of d-isomers of Met, Leu, Val, allo-Ile, Phe, Tyr, Trp and Arg. Tracer analysis using 14C-labeled d-Trp showed that d-Trp in the medium was used as a protein component in the cells, suggesting the presence of d-amino acid metabolic enzymes. Pyridoxal 5′-phosphate (PLP)-dependent racemase activity toward Met, Leu and Phe was detected in crude extract of P. horikoshii and was enhanced in cells grown in the medium supplemented with d-amino acids, especially d-allo-Ile. The gene encoding the racemase was narrowed down to one open reading frame on the basis of enzyme purification from P. horikoshii cells, and the recombinant enzyme exhibited PLP-dependent racemase activity toward several amino acids, including Met, Leu and Phe, but not Pro, Asp or Glu. This is the first report showing the presence in a hyperthermophilic archaeon of a PLP-dependent amino acid racemase with broad substrate specificity that is likely responsible for utilization of d-amino acids for growth.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pyrococcus horikoshii OT-3 is an anaerobic hyperthermophilic archaeon originally isolated from a hydrothermal vent in the Okinawa trough. Like other Pyrococcus strains, P. horikoshii generally uses complex proteinaceous media for growth (Gonzalez et al. 1998). These include peptone, tryptone and yeast extract, or a vitamin-supplemented amino acid mixture. However, in contrast to closely related strains such as P. furiosus and P. abyssi, P. horikoshii cannot grow on medium composed of casamino acids (acid hydrolysate of casein), which lacks Trp, Asn and Gln (Hoaki et al. 1993, 1994; Watrin et al. 1995). Cell growth can be achieved by supplementing medium based on casamino acids with Trp, indicating Trp to be an essential amino acid for the growth of P. horikoshii (Gonzalez et al. 1998).
In 1998, the genome of P. horikoshii was sequenced in the first total genome analysis of a hyperthermophilic archaeon (Kawarabayasi et al. 1998). Since then, the genomes of more than 170 archaeal strains have become available in the Kyoto encyclopedia of genes and genomes (KEGG) database (http://www.genome.jp/kegg/), which equipped with pathway maps for various metabolisms such as biosynthesis and degradation of amino acid. Typically, Trp is biosynthesized via the shikimate pathway, which starts with a condensation reaction between phosphoenolpyruvate and d-erythrose-4-phosphate and then ultimately produces chorismate, a common intermediate for the biosynthesis of not only Trp but also Phe and Tyr (Knaggs 2003). When we investigated the presence of genes involved in Trp biosynthesis in P. horikoshii using the KEGG database, none of genes in the gene cluster for Trp, Phe or Tyr biosynthesis via the shikimate pathway were present, which is consistent with the observed Trp requirement for P. horikoshii growth and suggests that P. horikoshii also requires Phe and Tyr for growth.
It is known that d-amino acids play important roles in various physiological processes in bacteria and eukarya, and are produced mainly by amino acid racemases (AARs), which catalyze racemization of amino acids (Yoshimura and Esaki 2003). It is also known that several archaeal strains accumulate d-amino acids such as d-Asp and d-Ser through the activities of Asp racemase (AspR) and SerR, respectively (Matsumoto et al. 1999; Nagata et al. 1999; Long et al. 2001; Ohnishi et al. 2008). Within the genome of P. horikoshii, three genes (PH0670, PH1054 and PH1733) have been annotated as racemase homologs. The PH0670 gene product was identified as a pyridoxal 5′-phosphate (PLP)-independent AspR, and its functional and structural properties have been characterized (Liu et al. 2002; Yoshida et al. 2006; Ohtaki et al. 2008). The structure of the protein encoded by PH1733 gene has been also determined as AspR-like protein, though the substrate for this enzyme is not Asp (Kita et al. 2009). And the structure of the PH1054 gene product has been determined (only deposited to Protein Data Bank; ID: 2eq5).
We recently found that P. horikoshii grows as well in casamino acid-based medium supplemented with d-Trp as it does in medium supplemented with l-Trp. This suggests that the d-Trp in the medium is incorporated into P. horikoshii cells and converted to l-Trp by an as-yet-unknown enzyme. In the present study, we investigated P. horikoshii’s requirement for each of 20 amino acids for growth and analyzed the corresponding genome information. In addition, we examined the effect of supplementing d-amino acids on growth and identified the enzyme responsible for d-amino acid utilization by P. horikoshii to be a novel AAR.
Materials and methods
Materials
d- and l-amino acids were purchased from Wako (Osaka, Japan) and Tokyo Chemical Industry (Tokyo, Japan). Horseradish peroxidase (HRP) and d-amino acid oxidase (DAO) from porcine kidney were purchased from Wako and Sigma-Aldrich (Tokyo), respectively. N-Ethyl-N-(2-hydroxy-3-sulfopropyl)-m-toluidine (EHSPT) and 4-aminoantipyrin (4-AA) were purchased from Dojindo (Kumamoto, Japan). Tryptone, yeast extract and casamino acids were purchased from BD Bioscience (Tokyo). Kao and Michayluk Vitamin Solution was purchased from Sigma-Aldrich. Side chain 3-14C-labeled d- and l-Trp (370 kBq each) were purchased from American Radiolabeled Chemicals (Saint Louis, MO, USA) via the Japan Radioisotope Association. Sequence grade-modified trypsin and ZipTip were purchased from Promega (Tokyo) and Merck Millipore (Tokyo), respectively, for matrix-assisted laser desorption ionization time-of-flight mass spectroscopy (MALDI TOF/MS). All other chemicals were of reagent grade.
Growth conditions of P. horikoshii
Medium containing tryptone and yeast extract was used as the standard medium for cultivating P. horikoshii OT-3 cells (Kawakami et al. 2004). For casamino acid-based medium, casamino acids (5 g/l) and Kao and Michayluk Vitamin Solution were used instead of tryptone and yeast extract, respectively, and the medium was supplemented with l-Trp (0.1 g/l). The defined medium (per L) consisted of 0.1 g each of 20 types of amino acids, 5 ml of vitamin solution (Robb and Place 1995), 25 g of NaCl, 1.3 g of (NH4)2SO4, 0.28 g of KH2PO4, 0.25 g of MgSO4·7H2O, 0.073 g of CaCl2·2H2O, 0.027 g of FeCl3·6H2O, 0.01 g of NaBr, 3.8 mg of Na2B4O7·10H2O, 2 mg of MnCl2·4H2O, 2 mg of (NH4)2Ni(SO4)2·6H2O, 0.29 mg of ZnSO4·7H2O, 0.17 mg of CuCl2·2H2O, 0.095 mg of AlK(SO4)2, 0.028 mg of CoSO4·7H2O, 0.024 mg of Na2MoO4·2H2O, 0.016 mg of VOSO4·2H2O, 0.016 mg of KI, 1 g of Na2S and 5 g of elemental sulfur. Resazurin was added to all media (1 mg/l) as an indicator of a reductive environment. The pH of all media was adjusted to 6.5–7.0 using NaOH. For growth, a seed culture (20 ml) was initially inoculated into the standard medium (200 ml) and cultivated at 90 °C without stirring (pre–pre-culture). After cultivation for 24 h, 10 % (v/v) of the culture was inoculated into a freshly prepared experimental medium (pre-culture) and cultivation was continued for 24 h. The same procedure was then repeated (main culture). After cultivation for 24–48 h, the cells grown in the main culture were cooled and collected by centrifugation (5000×g for 10 min), washed twice with 3 % NaCl, and used for experiments.
Incorporation of radiolabeled d-Trp into the cells
Radiolabeled Trp was used to confirm whether d-Trp in the medium was incorporated into P. horikoshii cells. About 370 kBq 14C-labeled d-Trp (corresponding to 37 μg) was added to 100 ml of main culture composed of casamino acid-based medium containing unlabeled d-Trp (0.01 g). After cultivation for 24 h, the cells were collected by centrifugation and washed three times with 3 % NaCl to remove any unincorporated isotope. After suspending the collected cells in 10 mM Tris/HCl (pH 7.5), aliquots of the suspensions were used for SDS-PAGE (Laemmli 1970). The gel was then evaporated onto filter paper, and the 14C-labeled Trp incorporated into proteins was analyzed using a bio-imaging analyzer BAS1000 (Fuji Film, Tokyo).
Detection of AAR activity
AAR activity was assessed based on the method of Sakuraba et al. (2002), which entailed detecting the quinoneimine dye produced by the condensation of 4-AA and EHSPT by HRP in the presence of H2O2 produced by the oxidation of a d-amino acid by DAO. The first reaction mixture (0.1 ml) for the racemase reaction contained 100 mM Tris/HCl (pH 8.0), 40 μM PLP, 10 mM l-amino acid and the enzyme. The reaction was started by adding the enzyme solution, after which the mixture was incubated at 80 °C for an appropriate time. The reaction was then stopped by cooling on ice for 10 min. For the production of quinoneimine dye, a second reaction mixture (0.1 ml) containing 100 mM Tris/HCl (pH 8.0), 0.4 mM 4-AA, 0.6 mM EHSPT, 0.02 U of DAO and 0.8 U of HRP was then added to the first reaction mixture and incubated at 37 °C for 30 min. Absorbance at 555 nm by the quinoneimine dye was measured using a U-1900 spectrophotometer (Hitachi, Tokyo) at room temperature. Under the assay conditions, the amount of d-amino acid produced by the racemase reaction was determined based on a calibration curve for each of d-amino acid at concentrations between 0 and 1.0 mM for Met, Leu and Pro, and between 0 and 2.0 mM for Ala, Val and Phe in the first reaction mixture. The activity was then expressed as the amount of d-amino acid (μmol) produced in 1 min at 80 °C.
Expression of target genes and enzyme purification
The expression plasmid harboring PH0670 gene (pET23a/PH0670) was a kind gift from Prof. Masafumi Yohda of Tokyo University of Agriculture and Technology. Three other genes, PH1054, PH1733 and PH0138, were amplified by PCR using P. horikoshii genomic DNA as a template, which was prepared as described previously (Kawakami et al. 2005). The primer sets used in the PCR reaction are listed in Supplementary Table S1. Forward and reverse primers introduced an NdeI site overlapping the 5′ initiation codon and a BamHI or BglII site proximal to the 3′ end of the termination codon. PCR reactions were performed using PrimeStar Max DNA polymerase (Takara, Tokyo) according to the manufacturer’s instructions. Amplified fragments were purified, introduced into pCR4-TOPO (Invitrogen, Tokyo) and sequenced. The resultant TOPO/PH1733 and TOPO/PH0138 were used as a template for silent mutations to disrupt the NdeI sites within the genes. To introduce the silent mutations, the sets of primers listed in Supplementary Table S1, whose sequences were designed to be complementary to each other, were used. After a non-PCR reaction using PrimeStar Max DNA polymerase, DpnI was added to the reaction mixtures to digest the template DNA, and plasmids with mutations were selected. The resultant TOPO/PH1733Silent, TOPO/PH0138Silent and TOPO/PH1054 were digested with NdeI and BamHI (BglII for PH0138) and introduced into pET15b or pET11a (Novagen, Tokyo) to generate pET15b/PH1054, pET15b/PH1733 and pET11a/PH0138, respectively.
Escherichia coli BL21 (DE3) CodonPlus RIPL cells (Stratagene, Tokyo) were transformed with pET23a/PH0670, pET15b/PH1054, pET15b/PH1733 or pET11a/PH0138, after which the transformants were cultivated in LB medium containing 100 μg/ml ampicillin at 37 °C for 4 h. Isopropyl-β-d-thiogalactopyranoside was then added to the medium to a final concentration of 0.5 mM, and cultivation was continued for an additional 3 h. The cells were then harvested by centrifugation, suspended in 10 mM Tris/HCl (pH 8.0) and disrupted by sonication. The resultant crude extract was heated at 80 °C for 20 min, and the supernatant was used to assay AAR activity.
To purify the recombinant PH0138, citrate buffer (pH 5.5) was added to the crude extract to 100 mM and heated at 90 °C for 60 min. After centrifugation, the supernatant was applied to Ether-Toyopearl (Tosoh, Tokyo), Butyl-Toyopearl (Tosoh) and DEAE-Cellulofine columns (Seikagaku Co., Tokyo), in that order, using the methods described below.
Purification of AAR from P. horikoshii and identification using MALDI TOF/MS
P. horikoshii cells (about 20 g wet weight) grown on standard medium were suspended in 10 mM Tris/HCl (pH 8.0) buffer and disrupted by sonication. The crude extract was collected as the supernatant after centrifugation (20,000×g for 15 min) and applied to a DEAE-Cellulofine column (about 100 ml) previously equilibrated with 10 mM Tris/HCl (pH 8.0). The column was then washed with the same buffer, and the enzymes were eluted using a linear gradient of 0–0.5 M NaCl in the same buffer. Active fractions were collected and ammonium sulfate was added to 40 % saturation. The resultant enzyme solution was applied to an Ether-Toyopearl column (about 100 ml) equilibrated with the buffer containing 40 % saturated ammonium sulfate, and the flow-through was continuously applied to a Butyl-Toyopearl column (about 50 ml) previously equilibrated with the same buffer. After washing the column with the same buffer, enzymes were eluted using a linear gradient of 40–0 % saturated ammonium sulfate. Active fractions were pooled and concentrated using a filter with a 30,000 molecular weight cut off (MWCO). The enzyme was applied to a Sephacryl S-200 (GE Healthcare, Tokyo) gel filtration column (about 300 ml) equilibrated with 10 mM Tris/HCl (pH 8.0) buffer containing 0.2 M NaCl, and eluted with the same buffer. Active fractions were collected and dialyzed against 10 mM Tris/HCl (pH 8.0).
The enzyme solution was next applied to an IEC DEAE-825 column (Shodex, Tokyo) previously equilibrated with 10 mM Tris/HCl (pH 8.0) on an HPLC system (Tosoh), and the enzymes were eluted in 1-ml fractions using a linear gradient of 0–0.25 M NaCl in the same buffer. The fractions for MS analysis were concentrated to 0.05–0.1 ml using an Amicon Ultra 0.5 with a 10,000 MWCO (Merck Millipore). The proteins were then separated by SDS-PAGE and stained with CBB R-250, after which the protein bands were cut from the gel, decolorized by incubation with 25 mM NH4HCO3 containing 50 % acetonitrile, reduced by incubation with 25 mM NH4HCO3 containing 10 mM DTT for 1 h at 56 °C, and alkylated by incubation with 25 mM NH4HCO3 containing 55 mM iodoacetamide for 45 min at room temperature. The protein was then digested in-gel with sequence grade-modified trypsin and was extracted as peptide fragments. After desalting using a ZipTip, the solution containing the peptide fragments was mixed with a matrix solution (saturated α-cyano-4-hydroxycinnamic acid dissolved in 0.1 % trifluoroacetic acid and 50 % acetonitrile), dried on a sample plate and analyzed using a 4700 MALDI TOF/TOF analyzer (AB SCIEX, Framingham, MA, USA). The obtained peptide mass data were then analyzed using the peptide mass fingerprinting method with Mascot search (Matrix Science, Boston, MA, USA).
Other experiments
Protein concentrations were determined using the Bradford method (1976), with bovine serum albumin serving as the standard. SDS-PAGE was carried out using the method of Laemmli (1970). Myosin (200 kDa), β-galactosidase (116.3 kDa), phosphorylase B (97.4 kDa), serum albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), trypsin inhibitor (21.5 kDa), lysozyme (14.4 kDa) and aprotinin (6.5 kDa) were used as molecular mass standards. For the separation and detection of free d- and l-amino acids within cells, an AccQ-Tag Ultra column (Waters, Tokyo) was used on an ultra-performance liquid chromatography (UPLC, Waters) system. The crude extracts from P. horikoshii cultivated under various conditions were treated with 10 % trichloroacetic acid to precipitate the proteins and then neutralized with NaOH. Free amino acids in the resultant solution were derivatized with o-phthaldialdehyde (OPA) and N-acetyl-l-cysteine or with OPA and N-tert-butyloxycarbonyl-l-cysteine for diastereomerization (Aswad 1984; Hashimoto et al. 1992; Mutaguchi et al. 2013). The derivatives were then analyzed using the AccQ-Tag Ultra column on the UPLC system, as described by Mutaguchi et al. (2013).
Results
Amino acid requirement and d-amino acid cultivation
To investigate the amino acid requirements of P. horikoshii, the cells were cultivated on defined medium in which one amino acid was omitted from the composition. Cell growth in medium from which Gly, Ala, Ser, Pro, Asn, Gln, Asp, Glu, Lys or Cys was omitted was similar to the growth observed in defined medium containing all 20 amino acids. On the other hand, the cells grew only slowly in medium lacking Met or Ile, and no cell growth was observed in medium lacking Thr, Leu, Val, Phe, Tyr, Trp, His or Arg (Table 1). P. horikoshii thus requires the presence of those eight amino acids in the medium for growth.
To examine utilization of d-amino acids during growth of P. horikoshii, the d-isomers of each of the eight amino acids required for growth or the two whose absence slowed growth were separately added to the defined medium instead of the l-isomers. In this case, d-allo-Ile (2R, 3S) and d-allo-Thr (2R, 3R) were used for cultivation as the d-isomers of Ile and Thr, respectively. P. horikoshii successfully grew in medium containing each of the d-isomers of Leu, Val, Phe, Tyr, Trp or Arg. Growth was also improved in medium supplemented with d-Met or d-allo-Ile (Table 1). However, no growth was observed in medium supplemented with d-allo-Thr or d-His. This suggests the eight effective d-amino acids were incorporated into P. horikoshii cells and utilized for growth.
Cultivation using radiolabeled d-amino acids
To confirm whether d-amino acids in the medium were incorporated into the cells during growth, free amino acids from within P. horikoshii cells cultivated with d-amino acids were analyzed using a UPLC system. However, no free d- or l-amino acids were detected, even in cells grown under the standard cultivation conditions, whereas free amino acids were readily detected in E. coli cells (data not shown).
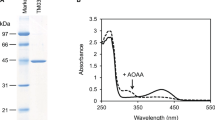
We next grew P. horikoshii cells in casamino acid-based medium supplemented with 14C-labeled d-Trp. After removing virtually all of the free 14C-labeled d-Trp from the cells by washing, 14C radioactivity was still clearly detected in the cell suspension, indicating that d-Trp was incorporated into the cells (data not shown). The same was observed with 14C-labeled l-Trp supplementation. After cell homogenates were subjected to SDS-PAGE, scanning the gel using an imaging analyzer revealed the presence of radioactivity in the proteins of cells cultivated with 14C-labeled d- or l-Trp (Fig. 1a, b). These results suggest that some of d-amino acids in the medium, including d-Trp, are incorporated into the cells and used as components of proteins.

SDS-PAGE of P. horikoshii cell homogenates (a) and autoradiography of the SDS-PAGE gel (b): lane 1 molecular markers; lanes 2–5 homogenates of cells cultivated on casamino acid-based medium containing unlabeled l-Trp (lane 2 about 20 μg), unlabeled d-Trp (lane 3 about 20 μg), unlabeled l-Trp and 14C-labeled l-Trp (lane 4 about 4 μg) and unlabeled d-Trp and 14C-labeled d-Trp (lane 5 about 4 μg)
Detection of AAR activity in the crude extract of P. horikoshii
We examined AAR activity in the crude extract of P. horikoshii as one of the enzyme reactions related to the metabolism of d-amino acids. To detect AAR activity, a spectrophotometric assay system was constructed as described in the Materials and methods section. To select detectable amino acids, the assays were performed using each of the eight d-amino acids (Pro, Met, Ser, Ala, Leu, allo-Ile, Val, and Phe) that exhibited more than 10 % relative activity (vs. d-Pro) in the DAO reaction (Tishkov and Khoronenkova 2005) in the first reaction mixture. It was found that the assay system was valid for detecting Met, Pro and Leu at concentrations between 0 and 1.0 mM and for detecting Ala, Val and Phe at concentrations between 0 and 2.0 mM (Supplementary Fig. S1a, b). d-Ser and d-allo-Ile were not detected with this assay system. When the first reaction was performed at 80 °C for 30, 60, 120 or 240 min in the presence of the dialyzed crude extract from P. horikoshii (72 μg of protein), color development caused by the produced d-amino acid was clearly detected in the reaction mixtures containing l-Met, l-Leu, l-Phe, l-Ala or l-Val as substrates, depending on the incubation times (Supplementary Fig. S1c). In the case of l-Pro, production of d-Pro did not depend on incubation time and the color development was slight, suggesting Pro was inert as a substrate. The specific activities of AAR in the crude extract after 60-min incubations were determined to be 0.0086, 0.0079, 0.0132, 0.0055 and 0.0032 μmol/min/mg for Met, Leu, Phe, Ala and Val, respectively. These results indicate that a novel AAR in the crude extract of P. horikoshii can catalyze the racemization of several amino acids. In the subsequent procedures for purification and gene identification of this enzyme, we mainly used Met and Leu as substrates in the assay because they yielded strong color development at low d-amino acid concentrations.
Effect of d-amino acid cultivation on AAR activity
To clarify the relationship between d-amino acid cultivation and AAR activity, we examined AAR activity in the extracts from cells cultivated under various conditions using Met as the substrate. The specific activities in the extracts prepared from cells grown in medium supplemented with a d-amino acid were increased 2–5 times, as compared to those grown in the standard medium (Fig. 2). In particular, extremely high specific activity (34 times higher) was detected in extract from cells cultivated with d-allo-Ile in the medium. This suggests expression of AAR genes is up-regulated by cultivation with d-amino acids, and the incorporated d-isomers are converted to the l-isomers by the AAR reaction.

AAR activity in crude extracts prepared from cells grown under various cultivation conditions. The crude extracts were prepared from the cells grown using standard cultivation (Std), l-amino acid cultivation (Laa) and d-amino acid cultivation with d-Met (Met), d-Leu (Leu), d-Phe (Phe), d-allo-Ile (Ile), d-Val (Val), d-Trp (Trp), d-Tyr (Tyr) or d-Arg (Arg). Thereafter, AAR activities in the crude extracts were assayed at 80 °C for 60 min using Met as the substrate. All assays were repeated three times and the average value was used
Purification and identification of the AAR
To identify the enzyme exhibiting the AAR activity detected in the crude extract, we first investigated the activity of three racemase homologs (PH0670, PH1054 and PH1733). These enzymes were successfully produced using an E. coli expression system and were highly purified by heat treatment. AAR activity was assessed using l-Met, l-Leu and l-Phe as substrates in a spectrophotometric assay; however, no AAR activity was detected from any of the three enzymes (data not shown).
We next tried purification of the enzyme exhibiting AAR activity from P. horikoshii cells and identification of the enzyme gene. The purification was carried out through five successive chromatography steps using DEAE-Cellulofine, Ether-Toyopearl, Butyl-Toyopearl, Sephacryl S-200 and IEC DEAE-825, but the enzyme was not highly purified, judging from SDS-PAGE (data not shown). We also tried using MALDI TOF/MS analysis to identify the gene encoding the AAR. After IEC DEAE-825 chromatography, AAR activity was detected in fraction No. 33, but not in No. 32. These fractions (1 ml each) were independently concentrated and subjected to SDS-PAGE (Fig. 3), after which we focused on the bands with molecular masses of about 50 and 27 kDa as the targets for MALDI TOF/MS analysis, because the density of those bands in fraction No. 33 was higher than that in No. 32. Peptide mass fingerprinting analysis revealed that peptide fragments from the 50 and 27 kDa proteins coincided with those derived from the gene product of PH0138 (coverage 50 %) and PH1733 (75 %), respectively. As mentioned above, the PH1733 gene product showed no AAR activity. The enzyme characteristics of the PH0138 gene product are still unknown, but it has already been annotated as a PLP-dependent 4-aminobutyrate aminotransferase (GABA-AT) with a molecular mass of 52,392. We therefore focused on the PH0138 gene product as a potential AAR.

SDS-PAGE of fractions from IEC DEAE-825 chromatography. Fraction numbers 32 and 33 were concentrated by filtration and applied to a 12 % gel. The left lane contains molecular weight markers
Recombinant PH0138 was successfully produced as a soluble protein in E. coli and remained in the soluble fraction after heat treatment at 90 °C for 60 min. Following the heat treatment, the recombinant enzyme was purified to homogeneity through three column chromatography steps. AAR activity toward Met, Leu and Phe was clearly detected in the purified enzyme, while no activity toward Pro was detected, confirming that PH0138 gene encodes an AAR. The specific activities of the purified enzyme were 10.5, 15.6, 22.7, 2.0 and 3.9 μmol/min/mg for Met, Leu, Phe, Ala and Val, respectively, indicating that Phe was the most preferred substrate among these amino acids.
It is known that racemases are generally divided into two groups, PLP-dependent and PLP-independent, and that the PLP is easily stripped from the enzyme during purification by ion exchange column chromatography and dialysis (Yoshimura and Esaki 2003). In the present study, the activity of the purified recombinant enzyme was still remaining in the absence of PLP (about 10 %) and fully restored by the addition of PLP. In addition, the activity could be completely blocked by addition of 1 mM hydroxylamine, a specific inhibitor of PLP-dependent enzymes.
Preliminary UPLC analysis was performed to investigate the enzyme’s substrate specificity (Supplementary Fig. S2). Using a d-amino acid as the substrate, the corresponding l-amino acid was clearly detected in reaction mixtures containing Met, Leu, Phe, Ala, Ser, Ile, Val, Trp or Tyr, and Asp and Glu were inert as substrates. These results indicate that PH0138 AAR is a broad substrate specificity amino acid racemase, BAR.
Discussion
This study was prompted by the observation that P. horikoshii can grow on medium containing d-Trp as the sole source of Trp. We found that for normal growth, P. horikoshii requires 10 amino acids in the medium, and that eight of those d-isomers serve as well as their l-isomers. In addition, we succeeded in identifying a novel BAR that is likely responsible for utilization of d-amino acids in this hyperthermophilic archaeon.
The amino acid requirements of several hyperthermophilic archaea, including Pyrococcus strains, have been previously investigated (Hoaki et al. 1993, 1994; Watrin et al. 1995). With the exception of the Trp requirement (Gonzalez et al. 1998), however, the amino acid requirements for growth of P. horikoshii had not been reported. In the present study, we found that the amino acid requirements of P. horikoshii are similar to those observed for P. abyssi GE5, which requires Thr, Leu, Ile, Val, Met, Phe, Tyr, His and Arg, but differ from those of P. furiosus DSM3638, which requires Ile and Val and grows weakly in the absence of Leu, Met or Thr (Hoaki et al. 1994; Watrin et al. 1995) (Table 1). This difference in amino acid requirements among P. horikoshii, P. abyssi and P. furiosus likely reflects the presence, or absence, of genes encoding the enzymes in each amino acid biosynthetic pathway. Analysis based on the KEGG database showed that P. horikoshii completely lacks the gene cluster for the shikimate pathway and the downstream pathways for biosynthesis of Trp, Phe and Tyr. Unlike P. horikoshii, both P. abyssi and P. furiosus possess the gene cluster for the shikimate pathway and downstream Trp biosynthetic pathway. However, the genes for biosynthesis of Phe and Tyr are missing from the gene cluster of P. abyssi, but are conserved in P. furiosus. These features are consistent with each strains’ respective aromatic amino acid requirements. Similarly, the differences in the requirements for His and Arg observed in these strains may be explained by the presence or absence of the corresponding gene cluster in their genomes. However, there are several contradictions between the experimental data and genome information. Although cultivation analysis suggests the presence of a biosynthetic pathway for Pro in these Pyrococcus strains, genome analysis indicates an absence of the genes for Pro biosynthesis. This suggests the presence of uncharacterized enzymes related to Pro biosynthesis in these hyperthermophilic archaea. Consistent with that idea, Cohen et al. (2003) pointed out that Pro biosynthesis in P. abyssi may proceed via a novel pathway. Conversely, P. horikoshii and P. abyssi showed a requirement for Thr, though the genes for Thr biosynthesis are completely conserved in their genomes, and growth in the absence of Thr was not improved by the addition of excess Asp, homoserine or Gly (data not shown). Asp and homoserine are the initial and intermediate products, respectively, in the Thr pathway, and Gly is converted to Thr via another pathway. It may be necessary to check the functions of the products encoded by the annotated genes.
We showed here that in the absence of the l-isomers, P. horikoshii is able to utilize d-isomers of Leu, allo-Ile, Val, Met, Phe, Trp, Tyr or Arg as protein components for growth (Table 1; Fig. 1). In addition, we identified a PLP-dependent BAR that catalyzes the racemization of a number of amino acids within P. horikoshii cells (Supplementary Figs. S1, S2). Notably, the BAR activity was enhanced in cells grown with d-amino acids, especially d-allo-Ile, as compared with cells grown on standard medium (Fig. 2). This suggests the identified BAR is responsible for utilization of d-amino acids for growth, though it is unclear why the enzyme is strongly up-regulated only when cultivated with d-allo-Ile. Further detailed analysis will be needed to fully understand the mechanism by which the gene is regulated.
We found that BAR is encoded by PH0138 gene, which was originally annotated as a GABA-AT. In addition, we found three or four GABA-AT homologs in Pyrococcus strains in the KEGG database, and these homologs are divided into four groups with more than 75 % sequence similarity (Table 2). Interestingly, the gene belonging to the group containing PH0138 is observed in the genomes of P. horikoshii, P. abyssi and Pyrococcus sp. NA2, but not in P. furiosus, P. yayanosii or Pyrococcus sp. ST04. Consistent with this finding, we were unable to experimentally detect BAR activity in the cell extract from P. furiosus (data not shown).
Recently, a PLP-dependent Ile 2-epimerase was identified in Lactobacillus buchneri JCM 1115 as a novel AAR (Mutaguchi et al. 2013). The Lactobacillus Ile 2-epimerase reacts with nonpolar amino acids such as Ile, Val and Leu as substrates, but not with Asp, Glu, Lys, Arg, His, Thr, Asn, Gln or Trp. Unlike the Lactobacillus Ile 2-epimerase, P. horikoshii BAR also reacts with Tyr and Trp, indicating that the P. horikoshii BAR has broader substrate specificity than the Lactobacillus Ile 2-epimerase. Like the P. horikoshii BAR, the gene encoding the Ile 2-epimerase was originally annotated as a GABA-AT. The primary structure of Lactobacillus Ile 2-epimerase (450 residues) showed 40, 37, 36 and 44 % homology with those of P. horikoshii BAR (466 residues), PH0782 (474 residues), PH1423 (454 residues) and PH1501 (438 residues), respectively, all of which are classified as fold type I PLP-dependent enzymes (Grishin et al. 1995; Mutaguchi et al. 2013). These observations prompted us to speculate that the three other GABA-AT homologs may also function as racemases or epimerases.
In this study, we identified a novel BAR that may be responsible for utilization of d-amino acids for growth in a hyperthermophilic archaeon. However, detailed functional and structural analyses of this BAR remain to be performed. Further investigations will be necessary to fully understand the role of BAR in d-amino acid metabolism.
References
Aswad DW (1984) Determination of d- and l-aspartate in amino acid mixtures by high-performance liquid chromatography after derivatization with a chiral adduct of o-phthaldialdehyde. Anal Biochem 137:405–409
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cohen GN, Barbe V, Flament D, Galperin M, Heilig R, Lecompte O, Poch O, Prieur D, Querellou J, Ripp R, Thierry JC, Van der Oost J, Weissenbach J, Zivanovic Y, Forterre P (2003) An integrated analysis of the genome of the hyperthermophilic archaeon Pyrococcus abyssi. Mol Microbiol 47:1495–1512
Gonzalez JM, Masuchi Y, Robb FT, Ammerman JW, Maeder DL, Yanagibayashi M, Tamaoka J, Kato C (1998) Pyrococcus horikoshii sp. nov., a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles 2:123–130
Grishin NV, Phillips MA, Goldsmith EJ (1995) Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci 4:1291–1304
Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T (1992) Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-l-cysteine and o-phthaldialdehyde. J Chromatogr 582:41–48
Hoaki T, Wirsen CO, Hanzawa S, Maruyama T, Jannasch HW (1993) Amino acid requirements of two hyperthermophilic archaeal isolates from deep-sea vents, Desulfurococcus strain SY and Pyrococcus strain GB-D. Appl Environ Microbiol 59:610–613
Hoaki T, Nishijima M, Kato M, Adachi K, Mizobuchi S, Hanzawa N, Maruyama T (1994) Growth requirements of hyperthermophilic sulfur-dependent heterotrophic archaea isolated from a shallow submarine geothermal system with reference to their essential amino acids. Appl Environ Microbiol 60:2898–2904
Kawakami R, Sakuraba H, Kamohara S, Goda S, Kawarabayasi Y, Ohshima T (2004) Oxidative stress response in an anaerobic hyperthermophilic archaeon: presence of a functional peroxiredoxin in Pyrococcus horikoshii. J Biochem 136:541–547
Kawakami R, Sakuraba H, Tsuge H, Goda S, Katunuma N, Ohshima T (2005) A second novel dye-linked l-proline dehydrogenase complex is present in the hyperthermophilic archaeon Pyrococcus horikoshii OT-3. FEBS J 272:4044–4054
Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Kikuchi H (1998) Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res 5:55–76
Kita A, Tasaki S, Yohda M, Miki K (2009) Crystal structure of PH1733, an aspartate racemase homologue, from Pyrococcus horikoshii OT3. Proteins 74:240–244
Knaggs AR (2003) The biosynthesis of shikimate metabolites. Nat Prod Rep 20:119–136
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Liu L, Iwata K, Kita A, Kawarabayasi Y, Yohda M, Miki K (2002) Crystal structure of aspartate racemase from Pyrococcus horikoshii OT3 and its implications for molecular mechanism of PLP-independent racemization. J Mol Biol 319:479–489
Long Z, Lee JA, Okamoto T, Sekine M, Nimura N, Imai K, Yohda M, Maruyama T, Sumi M, Kamo N, Yamagishi A, Oshima T, Homma H (2001) Occurrence of d-amino acids and a pyridoxal 5′-phosphate-dependent aspartate racemase in the acidothermophilic archaeon, Thermoplasma acidophilum. Biochem Biophys Res Commun 281:317–321
Matsumoto M, Homma H, Long Z, Imai K, Iida T, Maruyama T, Aikawa Y, Endo I, Yohda M (1999) Occurrence of free d-amino acids and aspartate racemases in hyperthermophilic archaea. J Bacteriol 181:6560–6563
Mutaguchi Y, Ohmori T, Wakamatsu T, Doi K, Ohshima T (2013) Identification, purification, and characterization of a novel amino acid racemase, isoleucine 2-epimerase, from Lactobacillus species. J Bacteriol 195:5207–5215
Nagata Y, Tanaka K, Iida T, Kera Y, Yamada R, Nakajima Y, Fujiwara T, Fukumori Y, Yamanaka T, Koga Y, Tsuji S, Kawaguchi-Nagata K (1999) Occurrence of d-amino acids in a few archaea and dehydrogenase activities in hyperthermophile Pyrobaculum islandicum. Biochim Biophys Acta 1435:160–166
Ohnishi M, Saito M, Wakabayashi S, Ishizuka M, Nishimura K, Nagata Y, Kasai S (2008) Purification and characterization of serine racemase from a hyperthermophilic archaeon, Pyrobaculum islandicum. J Bacteriol 190:1359–1365
Ohtaki A, Nakano Y, Iizuka R, Arakawa T, Yamada K, Odaka M, Yohda M (2008) Structure of aspartate racemase complexed with a dual substrate analogue, citric acid, and implications for the reaction mechanism. Proteins 70:1167–1174
Robb FT, Place AR (1995) Media for Thermophiles. In: Robb FT, Place AR (eds) Archaea-a laboratory manual-thermophiles. Cold Spring Harbor Laboratory Press, New York, pp 167–168
Sakuraba H, Satomura T, Kawakami R, Yamamoto S, Kawarabayasi Y, Kikuchi H, Ohshima T (2002) l-Aspartate oxidase is present in the anaerobic hyperthermophilic archaeon Pyrococcus horikoshii OT-3: characteristics and role in the de novo biosynthesis of nicotinamide adenine dinucleotide proposed by genome sequencing. Extremophiles 6:275–281
Tishkov VI, Khoronenkova SV (2005) d-Amino acid oxidase: structure, catalytic mechanism, and practical application. Biochemistry (Mosc) 70:40–54
Watrin L, Martin-Jezequel V, Prieur D (1995) Minimal amino acid requirements of the hyperthermophilic archaeon Pyrococcus abyssi, isolated from deep-sea hydrothermal vents. Appl Environ Microbiol 61:1138–1140
Yoshida T, Seko T, Okada O, Iwata K, Liu L, Miki K, Yohda M (2006) Roles of conserved basic amino acid residues and activation mechanism of the hyperthermophilic aspartate racemase at high temperature. Proteins 64:502–512
Yoshimura T, Esaki N (2003) Amino acid racemases: functions and mechanisms. J Biosci Bioeng 96:103–109
Acknowledgments
We are grateful to Mr. Yuji Kawahara and Ms. Kumi Yamashita for their excellent technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: J. Broos.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kawakami, R., Ohmori, T., Sakuraba, H. et al. Identification of a novel amino acid racemase from a hyperthermophilic archaeon Pyrococcus horikoshii OT-3 induced by d-amino acids. Amino Acids 47, 1579–1587 (2015). https://doi.org/10.1007/s00726-015-2001-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-2001-6