
Abstract
Camptothecin is a high-value anti-cancerous compound produced in many taxonomically unrelated species. Its biosynthesis involves a complex network of pathways and a diverse array of intermediates. Here, we report the functional characterization and regulation of secologanin synthase (NnCYP72A1), a cytochrome P450 involved in camptothecin biosynthesis from Nothapodytes nimmoniana. It comprises an open reading frame of 1566 bp in length. Heterologous expression in Saccharomyces cerevisiae and in vitro enzymatic assays using loganin as substrate confirmed the formation of secologanin. In planta transient overexpression analysis of NnCYP72A1 resulted in 4.21- and 2.73-fold increase in transcript levels of NnCYP72A1 on days 3 and 6 respectively. Phytochemical analysis of transformed tissues revealed ~ 1.13–1.43- and 2.02–2.86-fold increase in secologanin and CPT accumulation, respectively. Furthermore, promoter analysis of NnCYP72A1 resulted in the identification of several potential cis-regulatory elements corresponding to different stress-related components. Methyl jasmonate, salicylic acid, and wounding treatments resulted in considerable modulation of mRNA transcripts of NnCYP72A1 gene. Chemical analysis of elicitor-treated samples showed a significant increase in CPT content which was concordant with the mRNA transcript levels. Overall, the functional characterization and overexpression of NnCYP72A1 may plausibly enhance the pathway intermediates and serve as prognostic tool for enhancing CPT accumulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monoterpene indole alkaloids (MIAs) represent one of the largest and biologically most active class of alkaloids having diverse structural complexities and pharmacological activities (De Luca et al. 2014). This important category of metabolites is often produced via complex and highly regulated seco-iridoid pathway under the influence of different enzymatic reactions (Huang et al. 2016). The unique seco-iridoid pathway has been the target of many genetic engineering attempts, as it is an important source of potent terpenoid indole alkaloids. These primarily include reserpine, ajmalicine, ajmaline, serpentine, yohimbine, vinblastine, vincristine, quinine, and camptothecin (Pathania et al. 2016; Miettinen et al. 2014). However, the metabolic engineering of their biosynthetic networks in homologous/heterologous hosts has been greatly mired by poor understanding of their biosynthetic and regulatory mechanisms.
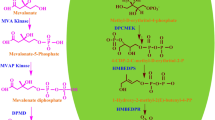
Nothapodytes nimmoniana is an important medicinal plant species endemic to Western Ghats of India. It is the richest source of quinolone indole alkaloid camptothecin (CPT) and 9 methoxy camptothecin (Ramesha et al. 2008; Rather et al. 2018). CPT is one of the most promising anticancer alkaloid with a potent DNA topoisomerase I inhibitory activity (Pommier 2009). Two water soluble semi-synthetic analogs topotecan and irinotecan derived from CPT exhibit enhanced pharmacological properties and clinical efficacy as compared to camptothecin and are widely used against the treatment of lung, breast, colorectal, cervical, and ovarian cancers (Isah and Mujib 2015). Despite the pharmacological relevance of camptothecin, its biosynthesis remains to be largely unresolved. However, transcriptome and proteome data of many alkaloid-producing plants like Catharanthus roseus (Apocynaceae), Ophiorhiza pumila (Rubiaceae), Camptotheca acuminata (Nyssaceae), Selaginella bryopteris (Selaginellaceae), and Salvia miltiorrhiza (Lamiaceae) provides a rich knowledge base for understanding the biosynthesis and regulatory components of plant-specialized metabolites, followed by molecular characterization and functional validation of the isolated candidate genes (Shukla et al. 2006; Sun et al. 2011; Yang et al. 2013; Yamazaki et al. 2013; Singh et al. 2018). Although, there have been concerted efforts over the last decade to decipher iridoid pathway but these investigations have led to the characterization of only few biosynthetic steps so far. Recently, transcriptomic analysis with untargeted metabolome profiling in O. pumila has facilitated the identification of a few more genes and possible intermediates involved in the CPT biosynthesis (Yamazaki et al. 2013). Cytochrome P450s are the important enzymes involved in the decoration of MIA pathway intermediates towards their end product (Giddings et al. 2011; Höfer et al. 2014; Salim et al. 2014). In N. nimmoniana, three unique NADPH cytochrome P450 reductases (CPRs) have been isolated and characterized, and their expressions examined in different organs (Huang et al. 2012). Transcriptome analysis of root and leaf tissues of N. nimmoniana led to the identification of large number of transcripts encoding for known and putative candidate genes involved in CPT biosynthesis, transcription factors, and many cytochrome P450 transcripts related to secondary metabolism (Rather et al. 2018). Nonetheless, most of the genes responsible for the biosynthesis of camptothecin are yet to be functionally characterized. With an objective to enhance the CPT production in N. nimmoniana via homologous modulation, we focused on the upstream steps of iridoid pathway (Fig. 1).

Putative camptothecin biosynthetic pathway: GES, geraniol synthase; G 8-H, geraniol 8-hydroxylase; CYP72A1, secologanin synthase; TDC, tryptophan decarboxylase; STR, strictosidine synthase. Double arrows represent the multiple steps between the intermediates
The seco-iridoid pathway diverges from the isoprenoid biosynthetic pathway at the intermediate geranyl diphosphate (GPP) (Pan et al. 2016). Geraniol synthase (GS) that has been characterized from C. acuminata hydrolyses GPP into geraniol which is regarded as the first committed step in seco-iridoid pathway (Chen et al. 2016). In the subsequent steps, geraniol undergoes hydroxylation, oxidation, reduction, glycosylation, and methylation reactions to produce secologanin which acts as a central compound from which ~ 2000 chemically diverse natural products arise (Irmler et al. 2000; Salim et al. 2014; Guirimand et al. 2010; Góngora-Castillo et al. 2012; Murata et al. 2008; Asada et al. 2013). It contains an aldehyde group suitable for condensation with the amino group of tryptamine to yield strictosidine, whose corresponding aglycone serves as the primary precursor for CPT biosynthesis (de Bernonville et al. 2015). However, the reaction steps between strictosidine and camptothecin are possibly catalyzed by cytochrome P450s but the specific reactions are yet to be deciphered (Mizutani 2012; Qu et al. 2015a, b).
During growth, development, and reproductive stages of an organism, gene regulation and expression events are precisely determined and controlled by promoters (Hernandez-Garcia and Finer 2014). In response to biotic and abiotic stresses, transcription factors interact with cis-regulatory elements of the promoters and activate the network of genes related to secondary metabolism (Biłas et al. 2016). For example, jasmonate (JA) regulated basic helix–loop–helix transcription factor triggers the expression of large number of genes that further catalyzes the sequential conversion of the ubiquitous terpenoid precursor geranyl diphosphate to the iridoid secologanin (Moerkercke et al. 2015; Cui et al. 2018. JA also activates the transcriptional regulator ORCA2, which has a role in regulating the terpenoid indole alkaloid (TIA) pathway (Li et al. 2013). Moreover, salicylic acid (SA), gibberellic acid, etc. elicitations have significantly improved the phytochemical constituents of Hypericum perforatum, Ocimum basilicum, Artemisia absinthium, and many other medicinally important plant species (Kim et al. 2006; Ali et al. 2015; Shakya et al. 2019). Wounding of plant tissues induces de novo synthesis of abscisic acid, JA, and ethylene which are known to activate a network of pathways for the enhancement of secondary metabolites as defense responses (Savatin et al. 2014). Elicitations in the form of phytohormones have emerged as effective means for the enhancement of high-value secondary metabolites in plant species. Therefore, it is of significant interest to identify critical cis-regulatory elements related to different abiotic and biotic stresses. The identified regulatory components may provide direct use to modulate secondary metabolism for enhanced metabolite production through application of elicitors and/growth regulators.
Overall, the chemical synthesis of structurally complex MIA compounds is costly, tedious, and impractical. Therefore, a deeper understanding of the regulatory system governing MIA metabolism is of particular interest for genetic and biotechnological interventions. Against this background, NnCYP72A1 was isolated and functionally validated. Moreover, its overexpression analysis resulted in the enhancement of transcript levels of NnCYP72A1 concomitant with higher accumulation of secologanin and CPT contents. Genome walking was employed to isolate upstream region of NnCYP72A1 for the identification of potential regulatory elements. Further, elicitation treatments were administered on the basis of identified cis-regulatory elements. Taken together, the present study provides useful insight into the robust functional efficacy and transcriptional regulation of NnCYP72A1.
Material and methods
Chemicals
β-Nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (NADPH) (CAS No. 2646-71-1) and camptothecin (CAS No. 7689-03-4) were purchased from Sigma-Aldrich (St. Louis, MO). Loganin (CAS No.18524-94-2) and secologanin (CAS No. 19351-63-4) were purchased from Santa Cruz Biotechnology, Inc. Longlife Zymolyase (Cat. No. 786-036) was purchased from G-Biosciences, USA. Organic solvents like methanol (CAS No. 67-56-1), acetonitrile (CAS No. 75-05-8), and formic acid (CAS No. 64-18-6) were of HPLC grade purchased from Merck (Darmstadt, Germany).
Plant material, RNA isolation, and cDNA synthesis
In vitro cultures of N. nimmoniana established at CSIR-Indian Institute of Integrative Medicine were used as a source material for experimental analysis. Total RNA was extracted by using SV Total RNA Isolation System (Promega, Madison, USA) as per manufacturer’s protocol. First-strand cDNA was synthesized by using Revert Aid First Strand cDNA Synthesis Kit (Thermoscientific, Lithuania) according to manufacturer’s protocol. Briefly, a 20 μL of reaction containing 3 μg of total RNA, 10 mM oligo (dT) primer, 10 mM dNTPs, 1 μL reverse transcriptase, and 2 μL of 10× buffer was incubated in thermal cycler (Eppendrof-22331, Hamburg, Germany) for 60 min at 42 °C followed by inactivation of reverse transcriptase by heating at 70 °C for 5 min.
Isolation and cloning of NnCYP72A1
The transcriptome resource of N. nimmoniana was explored to identify NnCYP72A1 transcripts displaying maximum homology with the characterized CYP72A1 genes of other iridoid producing plant species like O. pumila, C. roseus, and C. acuminata (Rather et al. 2018). RACE PCR strategy was used for amplification of remaining 5′ and 3′ ends of NnCYP72A1 using GeneRacer TM Kit according to the manufacturer’s protocol (Ambion, Austin, USA). Briefly, 5′ and 3′ RACE-ready first-strand cDNAs were synthesized from the total RNA of N. nimmoniana. The resultant RACE cDNAs were used for the amplification of 5′ and 3′ ends using RACE primers as listed in Supplementary file 1. First round of PCR amplification was performed with GeneRacer forward Primer-OUT with the reverse NnCYP72A1 RACE-OUT followed by a second round of PCR with NnCYP72A1 RACE-INN and GeneRacer nested primers. The PCR conditions are as follows: 1 cycle of 95 °C for 5 min and 35 cycles of 95 °C for 50 s, 50 °C for 30 s, 72 °C for 1:00 min with a final step at 72 °C of 5 min. The amplified products were ligated in pMD20-T vector (Mighty TA-cloning kit, Takara, Shiga 525-0058, Japan) and transformed into E. coli, DH5α host strain. Colonies were screened and plasmid from positive colonies was sequenced using M13 primers. Sequencing was performed with ABI PRISM 3130XL Genetic Analyser (Hitachi-high Technology Corporation Tokyo, Japan). The isolated 5′ and 3′ sequences were further aligned with partial putative sequence of NnCYP72A1 to generate full-length ORF of NnCYP72A1. Consequently, the full-length gene-specific primers (Supplementary file 1) were designed to amplify full coding sequence of NnCYP72A1 using a high-fidelity proof-reading DNA polymerase under following PCR program: 1 cycle of 95 °C for 5 min, 35 cycles of 94 °C for 30 s, 45 °C for 50 s, and 72 °C for 2 min followed by a final extension of 72 °C for 10 min. The resulted full-length ORF of NnCYP72A1 were cloned into pJET vector (Thermo Scientific, Vilnius, Lithuania), and transformed into E. coli, DH5α host strain. The transformed colonies were confirmed via colony PCR and further subjected to plasmid isolation using a DNA plasmid Miniprep Kit (Promega, Madison, WI, USA). The isolated plasmid was sequenced using M13 primers.
Bioinformatic analysis
The full-length nucleotide sequence of NnCYP72A1 was translated using the translate tool to predict ORF. Protein database (PDB) files were generated using Phyre2 server for predicting ligand-binding sites and three-dimensional structures of NnCYP72A1 (Kelley et al. 2015). ConSeq and ConSurf programs were used for the identification of functionally and structurally important residues in protein sequences (Ashkenazy et al. 2010). PyMOL tool (Schrödinger; http://www.pymol.org/) was used in the preparation of 3D protein structures. The ligand-binding sites were predicted using 3D Ligand Site server. The secondary structure was predicted by SOPMA program (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) and the properties of deduced amino acid residues were estimated using ProtParam (https://web.expasy.org/protparam/). In addition, signal sequence was determined by PHOBIUS program (Käll et al. 2007). Heme-iron ligand signature was identified in deduced protein sequence by using PROSITE server (http://prosite.expasy.org/cgi-bin/prosite). For phylogenetic analysis, open reading frame of NnCYP72A1 was searched in NCBI using BLASTp (http://www.ncbi.nlm.nih.gov/BLAST/) for retrieving homologous sequences, followed by multiple sequence alignment using the log-expectation (MUSCLE) alignment tool (http://www.ebi.ac.uk/Tools/msa/muscle) with the default parameters. The phylogenetic tree was constructed and visualized by MEGA7 software using neighbor-joining method with 1000 bootstrap replicates (Kumar et al. 2016).
Yeast expression
For heterologous expression in yeast, NnCYP72A1 gene was amplified with gene-specific expression primers (pYeDP60 SLS-KpnI; pYeDP60 SLS-EcoRI) harboring KpnI and EcoR1 restriction sites (Supplementary file 1). The amplified gene was cloned into pJET vector and plasmids were isolated from the positive colonies. The plasmid was digested with EcoR1 and Kpn1 enzymes and the construct was subcloned into pYeDP60 yeast expression vector. The expression construct was transformed into S. cerevisiae WAT11strain using LiCl method as described in (Pompon et al. 1996; Gietz and Schiestl 2007). Positive transformed yeast colony was grown on a selection medium at 30 °C and P450 expression was induced as described in (Höfer et al. 2013). Yeast cells were harvested by centrifugation and zymolase was added for digesting yeast cell walls along with manually breaking with glass beads of size 0.45 mm in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 600 mM sorbitol, and 1 mM EDTA. The homogenate was centrifuged for 10 min. at 10,000g. Pellet was discarded and the supernatant was again ultracentrifuged for 90 min. at 100,000g. Resultant supernatant was discarded and the pellet, comprising microsomal membranes, was resuspended in resuspension buffer containing 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 5% (v/v) glycerol and stored at − 80 °C. All procedures for microsomal isolation were performed at 0–4 °C and their cytochrome P450 content was measured by differential spectrophotometry according to Omura and Sato (Omura and Sato 1964). Microsomal protein was quantified using the Bradford colorimetric protein assay.
Enzymatic assay of NnCYP72A1
Standard enzymatic activities were carried out in 0.1 mL of 20 mM phosphate citrate buffer (pH 7.4) containing different substrate concentrations, 0.6 mM NADPH, and 0.5 mg of microsomal protein. After the addition of NADPH, samples were incubated at 30 °C for 30 min. The enzymatic reactions were quenched by acidification (10 μL of 20% HCl) and extracted with 200 μL ethyl acetate. Further, ethyl acetate extracts were evaporated to dryness and re-dissolved in methanol and samples were taken for LC-MS/MS analyses. Secologanin purchased from Santa Cruz Biotechnology (Delaware, California) was used as a reference standard for identification of NnCYP72A1-microsomal reaction products. Further, steady-state kinetic parameters were determined from initial velocity measurements where product formation was linear over the monitored time periods, using standard assay conditions with a fixed NADPH concentration (0.6 mM) and varied substrate concentration ranging from 50 to 300 μM. Formation of products was quantified and kinetic data of enzymatic reactions were fitted in Graph Pad Prism software and nonlinear regression analysis was used to calculate kinetic constant Km and Vmax of NnCYP72A1 protein.
Liquid chromatography mass spectrometry analysis
The Shimadzu LCMS-8030 (ESI) system (Tokyo, Japan), equipped with Hiber® HR RP-18 (50 × 2.1 mm inner diameter, 5 μm) column, was used for the analysis NnCYP72A1-microsomal reaction products. The mobile phase (A) consisted of 0.2% (v/v) acetic acid in methanol and mobile phase (B) 0.2% (v/v) acetic acid in water. For the detection of loganin, mobile phase (A) and (B) are used in the ratio of 50:50 (% v/v) and for secologanin 20:80 (% v/v), respectively. The column was operated at 30 °C with a flow rate of 0.5 mL/min. The injection volume of the sample was 10 μL. Mass spectrometer was operated in positive electrospray ionization (ESI) mode. Further, analysis of samples was performed in multiple reaction monitoring (MRM) mode using precursor/product ion transition at m/z 411.2/249.2 for secologanin and 413.0/219.3 for loganin. The interface temperature was maintained at 350 °C, drying gas flow 15 L/min and nebulizer gas flow 3 L/min. Total run time was of 3 min. The data obtained was analyzed using LabSolutions software.
Generation of overexpression construct
For in planta expression, NnCYP72A1 was amplified from cDNA using full-length gene-specific primers (Supplementary file 1) harboring NcoI and BglII enzyme restriction sites. PCR amplified product was digested with NcoI and BglII enzymes and further constructed in pCAMBIA1302 vector. NnCYP72A1-pCAMBIA1302 construct was transformed into Agrobacterium tumefacians (GV1301) strain. Overnight suspension culture of transformed A. tumefaciens was infiltered into leaves of N. nimmoniana as described by (Höfer et al. 2013). A. tumefacians transformed with empty vector was used as control. Agro-infiltrated plants were grown under controlled conditions until further analyses (16/8-h light/dark cycle, 28 ± 1 °C day, 25 ± 1 °C night). Leaves were harvested on days 3 and 6 of post-agroinfiltration for metabolic evaluation and transcriptional analysis to assess gene modulation. Experiments were performed in triplicates.
Genome walking and isolation of NnCYP72A1 promoter
Promoter sequence of NnCYP72A1 was isolated by the genome-walking method using the Genome Walker Universal Kit (Clontech). Briefly, primers (Supplementary file 1) were designed using the sequence information of cDNA of NnCYP72A1. Genomic DNA was isolated from leaf tissues of N. nimmoniana using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI 53711-5399 USA) according to the manufacturer’s protocol. For the construction of GenomeWalker DNA libraries, 5 μg of isolated genomic DNA was digested in four separate aliquots using four different restriction enzymes (EcoRV, DraI, PvuII, and StuI). Each set of digested samples were purified and ligated to the GenomeWalker AP adaptor provided with the kit to produce four adapter-ligated libraries. One microliter of each library was used separately as template in PCR amplification reaction. The primary PCR was performed using AP1 primer provided with kit and a gene-specific-out primer with following PCR conditions: 7 cycles at 94 °C for 25 s and 72 °C for 3 min; 35 cycles at 94 °C for 25 s, 67 °C for 3 min; and at 67 °C for 7 min. The product was diluted (1:50) and used in nested PCR, which was performed using AP2 provided with kit and gene specific-INN as nested primers under the following program: 5 cycles at 94 °C for 25 s and 72 °C for 3 min; 20 cycles at 94 °C for 25 s and 67 °C for 3 min, and followed by 67 °C for 7 min. The major PCR products were extracted, purified, and cloned into the pMD20-T cloning vector, transformed into E.coli, DH5α strain, and subsequently sequenced. The generated sequences were aligned with the 5′ region of their respective genes and further putative cis-regulatory elements upstream to start codon were identified by using PLACE (http://www.dna.affrc.go.jp/PLACE/), AtPAN (http://atpan.itps.ncku.edu.tw/index.php?id=promoter_analysis) and PlantCare (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) databases.
Elicitor treatments
To examine the effect of abiotic elicitors on the expression of NnCYP72A1, N. nimmoniana cultures grown at a growth chamber were subjected to different elicitor treatments in congruence with different motifs identified in promoter. Following elicitor treatments of methyl jasmonate (MeJA) (0.1 mM) and salicylic acid (SA) (0.1 mM), the samples were harvested at different time intervals (6 h, 12 h, 18 h, and 24 h). The harvested samples were further processed for RNA isolation and phytochemical (CPT) evaluation using HPLC. Some of the cultures were subjected to mechanical wounding by incising the leaves with a needle and the samples were harvested at different time intervals (6 h, 12 h, 18 h, and 24 h). The samples were further investigated at transcriptional and metabolic level for assessing the expression levels of NnCYP72A and CPT quantification. The untreated plant cultures were kept as control. The RNA isolated from the samples was reverse transcribed as discussed above. The effect of elicitor treatments on the expression of NnCYP72A1 was studied using quantitative real-time PCR (qRT-PCR).
Quantitative real-time expression
Total RNA was isolated from elicitor-treated and control samples of N. nimmoniana. About 1 μg of total RNA was used to synthesize the first-strand cDNA using Revert-aid Premium reverse transcriptase according to the manufacturer’s instructions. qRT-PCR analysis was carried out in triplicates using SYBR Green Universal Master Mix (Applied Biosystems, UK) in 48-well optical plates using ABI StepOne Real-time qPCR system (Applied Biosystems, USA). The standard 20 μL of reaction included 0.2 μL cDNA template, 200 nM each of the primers, and 10 μL SYBR Green mix under following cycling conditions: 1 cycle of 95 °C for 1 min, 40 cycle of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 20 s. Actin gene primers, taken as a normalization control were designed from the nucleotide sequence of Vitis vinifera (Accession No.: XM_010652725.2). All samples were analyzed in triplicates and the specificity of each primer pair was validated by a dissociation curve. The relative quantification method (2−ΔΔCT) was used to evaluate quantitative variation of NnCYP72A1 mRNA transcripts between the replicates (Livak and Schmittgen 2001).
Extraction and quantification of total camptothecin and secologanin contents by HPLC
Plant samples of N. nimmoniana were dried at room temperature and separately grounded into fine powder. Extraction of samples was performed as described by Fulzele and Satdive (2005), with slight modifications. Briefly, extracts of samples were prepared in methanol (3 × 50 mL; v/v) by stirring at room temperature (26 ± 2 °C) for 48 h. Further, the extracts were filtered and solvent was evaporated using rotary evaporator at 45 °C. For HPLC analysis, the extracts (10 mg/mL) were dissolved in HPLC grade methanol and filter sterilized with 0.45 μm membrane filters (Millipore, USA). The authentic standard camptothecin (1 mg/mL) purchased from Sigma-Aldrich and secologanin (1 mg/mL) from Santa Cruz Biotechnology were dissolved in HPLC grade methanol and used as the reference standard for detection and quantification. The HPLC system (Shimadzu CLASS-VP V 6.14 SPI model; Japan) equipped with RP-18e column (5 μm, 4.6× 250 mm; E-Merck, Bangalore, India), a quaternary gradient pump (LC-10AT VP model), and a PDA detector (SPD-M10A VP model) was used for the detection of CPT and secologanin in the samples. The analysis was performed by using mobile phase of acetonitrile: formic acid (99.5:0.5; v/v) which was delivered at a flow rate of 0.8 mL min−1. The injection volume of the samples was 10 μL and the column temperature was set at 30 °C to provide efficiency to the peaks. The detection wavelength set for UV chromatograms was 254 nm. The detection was made on the basis of retention time of reference standards camptothecin and secologanin under a set of specific column operating conditions as described above.
Results
Isolation and in silico analysis of complementary cDNA encoding NnCYP72A1
Using RACE PCR strategy, full-length coding sequence of NnCYP72A1 was isolated. The open reading frame (ORF) of NnCYP72A1 consisted of 1566 nucleotides which codes a polypeptide of 522 amino acids. The confirmed sequence was submitted to NCBI under accession no. KX129914. The homology BLASTx search of NnCYP72A1 in NCBI displays 77.41% sequence identity with O. pumila (Genbank ID: BAP90521.1), 77.01% with R. serpentina (GenBank ID: AGX93047.1), and 76% identity with C. roseus (AGX93064.1). The deduced amino acid sequence of NnCYP72A1 was found to have a calculated molecular weight of 59.8 kDa, having a cytochrome P450 cysteine heme-iron ligand signature sequence at 462–471 amino acid position with consensus sequence of FSWGPRVCLG. Three-dimensional protein model was determined using Phyre2 tool program (Fig. 2). Six templates were selected to model NnCYP72A1 protein based on heuristics to maximize confidence percentage identity using crystal structure of human CYP 11a in complex with 20, 22-2 dihydroxycholesterol. Homology modeling of NnCYP72A1 protein was performed with 90% confidence level. Four amino acids, which include Glu333, Thr334, Phe461, and Cys469, were found to be involved in the formation of catalytic pocket. Secondary structure analysis of NnCYP72A1 protein by SOPMA program (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) revealed that 50.86% protein is in α-helix form with extended strand (15.36%), beta turn (7.29%), and random coil (26.49%). 3D ligand-binding site web server results showed that 25 amino acids are involved in ligand-binding site rich in phenylalanine and leucine. Evolutionary conserved regions of NnCYP72A1 were determined by using Consurf programme http://consurf.tau.ac.il/ (Supplementary file 2). The well-known conservation of catalytically important residues of NnCYP72A1 sequences across the species was identified using the Clustal Omega tool https://www.ebi.ac.uk/Tools/msa/clustalo/. The conserved amino acid residues of P450s were reported to be preserved in the amino acid sequence of NnCYP72A1. Multiple sequence alignment displayed that NnCYP72A1 has maintained the conservation of functionally important regions with the CYP72A1 of Rauvolfia serpentina, Catharanthus roseus, Amsonia hubrichtii, Ophiorrhiza pumila, and C. acuminata, marked with green and yellow background (Supplementary file 2). Phylogenetic analysis was performed to ascertain the degree of evolutionary relationship of the deduced amino acid sequence of NnCYP72A1 with CYP72A1 of O. pumila (OpSLS: BAP90521.1), A. hubrichtii (AhSLS: AGX93046.1), C. roseus (CrSLS: AGX93064.1), R. serpentina (Rs SLS: AGX93047.1), Vinca minor (VM SLS: AGX93049.1), Tabernaemontana elegans (Te: KF415101.1), C. acuminata (Ca SLS: ADU19849.1). A total of 8 amino acid sequences of CYP72A1 were retrieved from NCBI (Fig. 2). The phylogenetic analysis revealed that CYP72A1 gene from different CPT-producing species was grouped in different clades. Furthermore, secologanin synthase of C. acuminata is highly deviated from other CPT-producing plants like O. pumila and N. nimmoniana. This divergence may be due to duplication events from unequal crossover during evolutionary process and that may possibly be the reason for production of secologanic acid instead of secologanin in C. acuminata (Magadum et al. 2013).

a–c Predicted three-dimensional models ligand-binding sites and phylogeny of NnCYP72A1: Three-dimensional (3D) protein model was determined by Phyre2 programs using the crystal structure of human CYP 11a1 in complex with 20,22-2 dihydroxycholesterol. N- and C-terminal domains are shown as red and pink caps, respectively. The catalytic and ligand-binding sites as predicted by 3DLigandSite web server, depicted in yellow and maroon color in the ribbon model and also highlighted as an inset respectively. The predicted amino acids involved in ligand-binding site of NnCY72A1 are ASP131, Val144, Trp152, Arg156,Phe163, leu327,Phe328, Ala331, Gly332, Thr335, Thr336, Leu339, Pro394, Val395, Leu398, Pro461,Phe462, Ser463, Arg467, Val 468, Cys469, Leu470, Gly471, Phe474, and Ala475. The phylogenetic analysis was performed using the MUSCLE program and MEGA7 software and the tree was reconstructed with neighbor-joining method. The numbers on the nodes indicate the bootstrap values after 1000 replicate. The analysis involved the alignment of 8 amino acid sequences that were chosen by analyzing the available data related to CYP72A1 genes from the NCBI database. The database accession numbers of the sequences used are as follows: N. nimmoniana secologanin synthase NnCYP72A1: KX129914), Ophiorrhiza pumila (OpSLS: BAP90521.1), Amsonia hubrichtii (AhSLS: AGX93046.1), Catharanthus roseus (CrSLS: AGX93064.1), Rauvolfia serpentine (Rs SLS: AGX93047.1), Vinca minor (VM SLS: AGX93049.1), Tabernaemontana elegans (Te: KF415101.1), and Camptotheca acuminata (Ca SLS: ADU19849.1)
Heterologous expression and functional validation of NnCYP72A1
To examine the catalytic function of NnCYP72A1, its ORF was transformed into pYeDP60 yeast expression vector and expressed under the control of galactose inducible promoter in S. cerevisiae strain Wat11 having P450-reductase ATR1 from A. thaliana. Induced cultures were grown at 30 °C and harvested after 18 h for microsome isolation as described in (Krithika et al. 2015). Microsomes of WAT11 yeast strain expressing NnCYP72A1 were incubated with loganin as a substrate for 30 min. at 30 °C. The reaction products were extracted and analyzed using LC-MS/MS in multiple reaction monitoring mode which revealed the formation of secologanin which was eluted at a retention time 0.65 min with ion transition at m/z 411.2 (Fig. 3). However, no activity was observed in yeast transformed with empty vector as control. Further, the purified microsomal proteins of NnCYP72A1 were used for investigating the kinetic properties. The amount of enzyme was kept constant whereas the concentration of the substrate was taken in increasing order (Supplementary file 3). The Vmax values of NnCYP72A1 for loganin, as calculated by nonlinear regression analysis, was 41.90 nmol min−1, whereas the apparent Km values was 20.80 μM (Fig. 4) as explained by Michaelis-Menten plot.

LC-MS/MS analysis of in vitro conversion of loganin to secologanin by the reaction of microsomes from yeast expressing NnCYP72A1. Multiple reaction monitoring (MRM) chromatogram and mass spectrometry spectra of substrate loganin which was eluted at about the retention time of 0.8 min with transition masses of m/z 413.0 (a, b); MRM and mass spectrometry spectra of reaction product secologanin which was eluted at about the retention time of 0.65 min with transition mass of m/z 411.2 (c, d) Chromatogram and mass spectra of authentic standard secologanin (e, f)

Kinetic study of NnCYP72A1. Michaelis-Menten plots of NnCYP72A1. The kinetic parameters Km and Vmax were calculated by nonlinear regression analysis using GraphPad Prism 6 software. The activity of NnCYP72A1 was assayed in 20 mM citrate phosphate buffer. The loganin was used as substrate, and the production of secologanin was quantified as activity (nmol/min)
Overexpression of NnCYP72A1
Using transient expression system, NnCYP72A1 was overexpressed in the leaves of N. nimmoniana to predict its role in CPT biosynthesis. The fusion construct of NnCYP72A1 was prepared in pCAMBIA1302 plant expression vector at the 5′-terminal end of the green fluorescent protein (GFP) gene under the control of CaMV 35S promoter (Fig. 5a). The overexpression construct of NnCYP72A1-pCAMBIA 1302 (OE) and empty vector (EV) were transformed into host plant via agroinfiltration and expression was confirmed via green fluorescent microscopy under 450-nm wavelength (Olympus SN-3M08785, Tokyo, Japan) (Fig. 5b–g). Samples were harvested after days 3 and 6 of post-agroinfiltration and investigated at molecular level by assaying NnCYP72A1 modulation as well as at metabolic level by quantifying secologanin and CPT contents. qRT-PCR analysis of NnCYP72A1 in transformed leaves showed 4.21-fold increase in transcript levels on day 3 and 2.73-fold increase on day 6 in comparison to non-transformed control (Fig. 5h). A slight increase of NnCYP72A1 expression was also observed in leaves transformed with empty vector. Interestingly, phytochemical analysis of transformed leaf showed 1.43- and 1.13-fold increase in secologanin accumulation on days 3 and 6, respectively (Fig. 5i), while CPT content was increased to 2.86- and 2.02-fold on days 3 and 6, respectively (Fig. 5j).

Overexpression of NnCYP72A1. Construct of NnCYP72A1 in pCAMBIA1302 vector at the 5′-terminal end of the green fluorescent protein (GFP) gene under the control of CaMV 35S promoter (a); expression analysis of transformed leaves under normal light (b, e); transformed leaves under green fluorescence microscope under wavelength 450 nm (Olympus SN-3M08785, Tokyo, Japan) (c, f); merged image of b and c (d); and merged image of e and f (g), bars = 50 μm; qRT-PCR analysis of NnCYP72A1 in control (c), leaves transformed with empty vector (EV) and leaves transformed with overexpression construct NnCYP72A1(OE) on days 3 and 6 of post-agroinfiltration (h); determination of secologanin and camptothecin accumulation in transformed leaves in comparison to control (i, j). Values are expressed as means ± SD representing three independent biological samples, each with three technical replicates. Differences were scored as statistical significance at **P < 0.01 levels and *P < 0.05. Asterisks indicate difference in expression levels of genes in transformed and control plants
Tissue-specific transcript profile
To understand the spatial regulation of the NnCYP72A1, the expression analysis of NnCYP72A1 was carried out in different tissues like leaf, young stem, old stem, and root of N. nimmoniana using qRT-PCR. The transcript levels of CYP72A1 were detected in all the examined tissues, but their levels were observed to be differential. qRT-PCR analysis of NnCYP72A1 showed the highest levels of transcripts in root, followed by leaf, young stem, and old stem (Fig. 6).

Tissue-specific real-time expression analysis. Quantitative assessment of the expression of NnCYP72A1 in leaf, young stem, old stem, and root of Nothapodytes nimmoniana. β-Actin was used as an endogenous control to normalize the expression of NnCYP72A1. The data were compared and analyzed with one-way ANOVA using GraphPad Prism 6 software. Values are means ± SD of three technical replicates indicated by bars. Differences were scored as statistical significance at **P < 0.01 and *P < 0.05 levels. Asterisks indicate the comparison of expression levels of NnCYP72A1 in leaf with other plant parts
Isolation and in silico characterization of promoter
To elucidate the potential regulators involved in transcriptional regulation of NnCYP72A1 gene, its promoter was isolated and predicted for the presence of various putative cis-regulatory elements using PLACE, ATPAN, and PlantCare databases. Using PCR-based genome-walking strategy, a 567-bp promoter region of NnCYP72A1 gene was isolated. The transcription initiation site (TIS) was determined by 5′ RACE analysis and is located at 46-bp upstream of the ATG start codon in NnCYP72A1 (Supplementary file 4). Putative TATA box of NnCYP72A1 was located at 32 bp upstream of the TIS site. Several important cis-regulatory elements involved in gene regulation were identified within the promoter regions of NnCYP72A1. These included hormone-responsive, light-responsive, and various other stress-related elements (Table 1). Among these cis-regulatory elements, TGACG motif was selected to investigate their role in elicitor responsiveness with an aim to study the inducible/repressible nature of regulatory motifs.
Elicitor treatment
The recent advances in understanding the plant signaling pathways have paved way for using elicitor induced modulation of plant secondary metabolism as a predictive tool for pathway engineering and regulation-based studies. The elicitors chosen on the basis of promoter analysis were evaluated with regard to NnCYP72A1 expression pattern vis-à-vis CPT accumulation to understand their regulatory role. The effect of wounding and two elicitors, namely MeJA (0.1 mM) and SA (0.1 mM), on expression of NnCYP72A1 was investigated at 6-h, 12-h, 18-h, and 24-h time intervals. The results showed that the expression pattern NnCYP72A1 was strongly increased by phytohormone treatment, though the expression levels were different for different elicitors. MeJA enhances the expression levels of NnCYP72A1 to ~ 0.6–1.6-fold as compared to control (Fig. 7a). Further, the effect of MeJA was also monitored at metabolite level by assaying CPT content (Supplementary file 5). The HPLC analysis of MeJA-treated samples showed 0.73- to 2.2-fold increase in CPT content as compared to control (Fig. 7b). The widely distributed phytohormone SA plays an important role in plant defense reactions against a broad range of stresses through morphological, physiological, and biochemical mechanisms. After SA treatment, the expression levels of NnCYP72A1 were increased by 0.43–1.27-fold (Fig. 7c). Further, SA treatment also demonstrated 0.64–1.37-fold increase in CPT content in comparison to control (Fig. 7d). Mechanical stresses like wounding produce endogenous signaling cascade which are mediated by hormones such as JA, ethylene, SA, and ABA. Further, upon wounding, the response was somewhat vigorous due to volatile JA released from wounded tissues that resulted in 0.44–1.23-fold increase in mRNA transcript levels of NnCYP72A1 as compared to control (Fig. 7e). Moreover, phytochemical analysis revealed 0.49–1.93-fold increase in CPT content as compared to control (Fig. 7f) (Supplementary file 5). Overall, MeJA, SA, and wounding significantly enhanced the expression level of NnCYP72A1 which was confirmed both at transcriptional level as well as at metabolite level.

Time course effect of elicitor treatments on expression profiles of NnCYP72A1. Time course effect on expression of NnCYP72A1 and camptothecin accumulation pattern in micropropagated Nothapodytes nimmoniana in response to elicitations by 0.1 mM MeJA (a, b), 0.1 mM SA (c, d), and wounding (e, f). The in vitro raised cultures were elicited and harvested after different time intervals. Actin was used as an endogenous control. The data were compared and analyzed with one-way ANOVA using GraphPad Prism 6 software. Values represented as means ± SD of three independent biological replicates each with three technical replicates. Differences were scored as statistical significance at **P < 0.01 levels and *P < 0.05. Asterisks indicate difference in expression levels of genes in elicitor-treated and control plants
Discussion
Monoterpene Indole alkaloids are a large group of plant-derived natural bioactive products. Their biosynthesis is a complex process with more than 50 biosynthetic steps comprising the large array of genes, enzymes, regulators, and transporters (Zhao et al. 2013). The structural diversity, low production, and diverse functions of MIAs have made them obvious targets for genetic engineering to enhance their production (Leonard et al. 2009). The pivotal pharmacological significance of MIAs prompted us to target the penultimate gene of seco-iridoid pathway, i.e., secologanin synthase (NnCYP72A1). It catalyzes the conversion of loganin into secologanin. Its full-length ORF was cloned using transcriptome resource of N. nimmoniana whose amino acid sequence is 77% similar to O. pumila and R. serpentina. Seco-iridoid pathway has been extensively studied in many alkaloid-producing plants in which methyl esters of loganin and secologanin are used as intermediates in the biosynthesis of strictosidine, a universal precursor of all MIAs (Irmler et al. 2000; Höfer et al. 2013; Yamazaki et al. 2013). Recently, metabolic profiling of C. acuminata tissues revealed the presence of loganic acid and secologanic acid instead of loganin and secologanin. These intermediates are involved in the biosynthesis of strictosidinic acid (Sadre et al. 2016). It was therefore tempting to validate the catalytic activity of CYP72A1 by in vitro enzymatic assay. Microsomes isolated from transformed yeast containing NnCYP72A1 efficiently converted loganin into secologanin as confirmed by mass spectrometry and fragmentation patterns. Further, efficiency of NnCYP72A1 towards its substrate was investigated using enzyme kinetics (Fig. 4). The kinetic characterization of NnCYP72A1 determined its catalytic efficiencies with an apparent Km of 20.80. Overall, it confirms secologanin as intermediate of CPT pathway operating in N. nimmoniana as reported earlier by the authors (Rather et al. 2018).
Secondary metabolism of plants is strictly controlled by genes of the biosynthetic machinery; one way to enhance the yield is to regulate the key pathway genes. For example, MIA levels of C. roseus have been improved by transient overexpression of geraniol synthase and geranyl geranyl diphosphate synthase (Kumar et al. 2018). Sharma et al. (2018) established transgenic lines of C. roseus in which 2-fold increase of vinblastine, vindoline, and catharanthine alkaloids has been observed due to overexpression of tryptophan decarboxylase and strictosidine synthase genes of seco-iridoid pathway. However, there are no reports for in planta overexpression of CYP72A1. Therefore, transient overexpression of NnCYP72A1 was performed to investigate its effect on secondary metabolism of N. nimmoniana. Overexpression of NnCYP72A1 resulted in significant enhancement in its mRNA transcript levels. The qRT-PCR analysis of transformed leaves showed ~ 2.73–4.21-fold increase of NnCYP72A1 mRNA transcript levels. These results were further corroborated with metabolite accumulation by assaying secologanin and CPT contents. The HPLC analysis revealed higher accumulation of secologanin and CPT concentrations (~ 1.43- and 2.86-fold respectively) (Fig. 5). These results conceivably indicate that the enhanced availability of upstream pathway precursors may be augmenting the production of CPT in N. nimmoniana. An advance in computational techniques to predict protein structure and function is becoming an important tool for successful metabolic engineering (Höfer et al. 2013). Molecular docking and homology modeling studies of proteins provide an insight into the acceptor and donor specificities of enzymes and combination of different bioinformatic tools can help to predict more information related to their functions (Bhat et al. 2013). Bioinformatic analysis of NnCYP72A1 was carried out for predicting the ligand-binding and catalytic sites. The presence of iron-binding signature is a characteristic feature of cytochromes and signal sequence has been identified to substantiate its functionality at bioinformatic level. CYP72 clan is one of the largest groups that contributes to secondary metabolism in plants. This clan has been diverged swiftly, but each lineage of the clan appears to have evolved species-specific functions (Prall et al. 2016). CPT is produced in several unrelated orders of angiosperms and it has been hypothesized that genes of CPT biosynthetic pathway have evolved during early evolution. The CPT pathway genes might have been switched off for a certain period of time and recuperate function at some later stage during the evolutionary process (Lorence and Nessler 2004). In this study, phylogenetic analysis of NnCYP72A1 was performed with other characterized CYP72A1 to predict its evolutionary relationship. Phylogenetic analysis of NnCYP72A1 showed its divergence from other alkaloid-producing plants but it retains orthologous nature with O. pumila, C. roseus, and R. serpentina. However, CYP72A1 of C. acuminata is highly diverged from other seco-iridoid-producing species and in C. acuminata, it produces secologanic acid instead of secologanin as an intermediate of seco-iridoid pathway. Hence, phylogenetic relationship of NnCYP72A1 also establishes its close relationship with the CYP72A1 of secologanin-producing plant species.
Studies of gene promoters that regulate gene expression at the transcriptional level are vital for basic understanding of transcriptional targeting and gene regulation (Hernandez-Garcia and Finer 2014). The detailed investigation of cis-regulatory elements along with their corresponding transcription factors provide basic understanding about the controlling, activation and suppression of gene expression when subjected to diverse environmental and extracellular factors (Dhar et al. 2014). In our study, NnCYP72A1 promoter contained several eukaryotic cis-regulatory elements, including CAAT and TATA boxes, TGACG motif, and WRKY (Table 1). These identified cis-acting elements are related to tissue specificity, drought stress, light-controlling elements, and phytohormones. Further, elicitations were chosen on the basis of these cis-regulatory elements to gain an insight into regulation of NnCYP72A1 and corroborate their inducible/repressible nature. From previous studies, methyl jasmonate plays a crucial role in regulating defensive and developmental genes, enhancing transcriptional reprogramming of secondary metabolism, concerted upregulation of genes involved in specific specialized biosynthetic pathways (De Geyter et al. 2012). In Ocimum basilicum and Coffea canephora, various concentrations (0.1–0.5 mM) of MeJA and SA were used for the transcriptional regulation of genes (Kim et al. 2006; Kumar and Giridhar 2015). In the present investigation, MeJA (0.1 mM) elicitation enhanced the expression levels of NnCYP72A1 to ~ 0.6–1.6-fold as compared to control (Fig. 7a). Further, chemoprofiling of the samples harvested at different time periods were found to be concordant with the expression pattern of NnCYP72A1. These results further confirm the responsiveness of MeJA motif in the promoter region of NnCYP72A1 as deciphered through in silico analysis. Moreover, our results are in good agreement with earlier studies wherein the expression level of CYP94C1 was enhanced by treatment with methyl jasmonate and wounding (Kandel et al. 2007). In C. roseus, it has been demonstrated that MeJA treatment has profound effect on the regulation of STR and TDC via ORCA3 and results in increased accumulation of terpenoid indole alkaloids (Peebles et al. 2009). The exogenous treatment of plants with SA induces the metabolic changes that involve discerning modulation of gene expression as well as enhanced metabolite accumulation (Pandith et al. 2016). Additionally, previous reports have shown that wounding, MeJA, and SA elicitation treatments are potential factors for the alteration of transcriptional levels of various genes that in due course affect secondary metabolite production (Peebles et al. 2009; Ali et al. 2007). In the present study, SA treatment of N. nimmoniana enhanced the expression levels of NnCYP72A1 in the range of 0.43–1.27-fold and HPLC analysis of SA-treated samples demonstrated 0.64–1.37-fold increase in CPT content in comparison to control (Fig. 7d). The wounding of plants results in production of oligogalacturonides and peptides, like systemin. These substances immediately induce ethylene and jasmonic acid responsive genes and other genes related to oxidative stress, heat-shock proteins, dehydration stress, etc. Wounding-induced genes encode proteins involved in signal transduction, such as members of AP2, MYB, and WRKY families (Cheong et al. 2002). It was evaluated by inflicting wounding of N. nimmoniana leaf tissues. Consequent effects were monitored by assaying NnCYP72A1 expression and further quantifying CPT concentration. The results showed that wounding enhances NnCYP72A1 transcript levels in the range 0.44–1.23-fold. It was reflected in the higher CPT accumulation (Fig. 7a–f). Overall, these results confirm the presence of different stress regulating motifs in the promoter of NnCYP72A1. Therefore, phytohormone-mediated elicitations would be a viable alternative to accelerate gene expression and enhance the production of seco-iridoid metabolites.
In conclusion, NnCYP72A1 of seco-iridoid pathway has been functionally characterized in heterologous host yeast. LC-MS/MS confirmed its functionality which was further corroborated by bioinformatic analysis. Moreover, its overexpression studies triggered copious transcriptional activity resulting in higher accumulation of secologanin and CPT concentrations. Additionally, isolation and characterization of promoter led to the identification of several important upstream cis-regulatory components. Further, elicitor-mediated expression analysis provided better insight regarding the regulation of NnCYP72A1. Taken as a whole, functional characterization, overexpression analysis, and transcriptional regulation of NnCYP72A1 in relation to enhanced CPT concentration may be further exploited for homologous modulation via genetic engineering.
References
Ali MB, Hahn EJ, Paek KY (2007) Methyl jasmonate and salicylic acid induced oxidative stress and accumulation of phenolics in Panax ginseng bioreactor root suspension cultures. Molecules. 12:607–621
Ali M, Abbasi BH, Ali GS (2015) Elicitation of antioxidant secondary metabolites with jasmonates and gibberellic acid in cell suspension cultures of Artemisia absinthium L. Plant Cell Tissue Organ Cult 120:1099–1106
Asada K, Salim V, Masada-Atsumi S, Edmunds E, Nagatoshi M, Terasaka K, Mizukami H, De Luca VA (2013) 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 25:4123–4134. https://doi.org/10.1105/tpc.113.115154
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:529–533
Bhat WW, Dhar N, Razdan S, Rana S, Mehra R, Nargotra A, Dhar RS, Ashraf N, Vishwakarma R, Lattoo SK (2013) Molecular characterization of UGT94F2 and UGT86C4, two glycosyltransferases from Picrorhiza kurrooa: comparative structural insight and evaluation of substrate recognition. PLoS One 8(9):e73804. https://doi.org/10.1371/journal.pone.0073804
Biłas R, Szafran K, Hnatuszko-Konka K, Kononowicz AK (2016) Cis-regulatory elements used to control gene expression in plants. Plant Cell Tissue Organ Cult 127:269–287
Chen F, Li W, Jiang L, Pu X, Yang Y, Zhang G, Luo Y (2016) Functional characterization of a geraniol synthase-encoding gene from Camptotheca acuminata and its application in production of geraniol in Escherichia coli. J Ind Microbiol Biotechnol 43:1281–1292
Cheong Y, Chang HS, Gupta R, Wang X, Zhu T, Luan S (2002) Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiol 129:661–677
Cui X, Wang YX, Liu ZW, Wang WL, Li H, Zhuang J (2018) Transcriptome-wide identification and expression profile analysis of the bHLH family genes in Camellia sinensis. Funct Integr Genomics 18:489–503. https://doi.org/10.1007/s10142-018-0608-x
de Bernonville TD, Foureau E, Parage C, Lanoue A, Clastre M, Londono MA, Oudin A, Houillé B, Papon N, Besseau S, Glévarec G (2015) Characterization of a second secologanin synthase isoform producing both secologanin and secoxyloganin allows enhanced de novo assembly of a Catharanthus roseus transcriptome. BMC Genomics 16:619. https://doi.org/10.1186/s12864-015-1678-y
De Geyter N, Gholami A, Goormachtig S, Goossens A (2012) Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci 17:349–359
De Luca V, Salim V, Thamm A, Masada SA, Yu F (2014) Making iridoids/secoiridoids and monoterpenoid indole alkaloids: progress on pathway elucidation. Curr Opin Plant Biol 19:35–42. https://doi.org/10.1016/j.pbi.2014.03.006
Dhar N, Rana S, Razdan S, Bhat WW, Hussain A, Dhar RS, Vaishnavi S, Hamid A, Vishwakarma R, Lattoo SK (2014) Cloning and functional characterization of three branch point oxidosqualene cyclases from Withania somnifera (L.) dunal. J Biol Chem 289:17249–17267
Fulzele DP, Satdive RK (2005) Comparison of techniques for the extraction of the anti-cancer drug camptothecin from Nothapodytes foetida. J Chromatogr 1063:9–13. https://doi.org/10.1016/j.chroma.2004.11.020
Giddings LA, Liscombe DK, Hamilton JP, Childs KL, DellaPenna D, Buell CR, O’Connor SE (2011) A stereoselective hydroxylation step of alkaloid biosynthesis by a unique cytochrome P450 in Catharanthus roseus. J Biol Chem 286:16751–16757
Gietz RD, Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34
Góngora-Castillo E, Childs KL, Fedewa G, Hamilton JP, Liscombe DK, Magallanes-Lundback M, Mandadi KK, Nims E, Runguphan W, Vaillancourt B, Varbanova-Herde M (2012) Development of transcriptomic resources for interrogating the biosynthesis of monoterpene indole alkaloids in medicinal plant species. PLoS One 7:e52506. https://doi.org/10.1371/journal.pone.0052506
Guirimand G, Courdavault V, Lanoue A, Mahroug S, Guihur A, Blanc N, Giglioli-Guivarc’h N, St-Pierre B, Burlat V (2010) Strictosidine activation in Apocynaceae: towards a “nuclear time bomb”? BMC Plant Biol 10:182. https://doi.org/10.1186/1471-2229-10-182
Hernandez-Garcia CM, Finer JJ (2014) Identification and validation of promoters and cis-acting regulatory elements. Plant Sci 217:109–119
Höfer R, Dong L, André F, Ginglinger JF, Lugan R, Gavira C, Grec S, Lang G, Memelink J, Van Der Krol S, Bouwmeester H (2013) Geraniol hydroxylase and hydroxygeraniol oxidase activities of the CYP76 family of cytochrome P450 enzymes and potential for engineering the early steps of the (seco) iridoid pathway. Metab Eng 20:221–232
Höfer R, Boachon B, Renault H, Gavira C, Miesch L, Iglesias J, Ginglinger JF, Allouche L, Miesch M, Grec S, Larbat R (2014) Dual function of the cytochrome P450 CYP76 family from Arabidopsis thaliana in the metabolism of monoterpenols and phenylurea herbicides. Plant Physiol 166:1149–1161
Huang FC, Sung PH, Do YY, Huang PL (2012) Differential expression and functional characterization of the NADPH cytochrome P450 reductase genes from Nothapodytes foetida. Plant Sci 190:16–23
Huang Y, Tan H, Guo Z, Wu X, Zhang Q, Zhang L, Diao Y (2016) The biosynthesis and genetic engineering of bioactive indole alkaloids in plants. J Plant Biol 59:203–214
Irmler S, Schröder G, St-Pierre B, Crouch NP, Hotze M, Schmidt J, Strack D, Matern U, Schröder J (2000) Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J 24:797–804
Isah T, Mujib A (2015) Camptothecin from Nothapodytes nimmoniana: review on biotechnology applications. Acta Physiol Plant 37. https://doi.org/10.1007/s11738-015-1854-3
Käll L, Krogh A, Sonnhammer EL (2007) Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res 35:429–432
Kandel S, Sauveplane V, Compagnon V, Franke R, Millet Y, Schreiber L, Werck-Reichhart D, Pinot F (2007) Characterization of a methyl jasmonate and wounding responsive cytochrome P450 of Arabidopsis thaliana catalyzing dicarboxylic fatty acid formation in vitro. FEBS J 274:5116–5127
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858
Kim HJ, Chen F, Wang X, Rajapakse NC (2006) Effect of methyl jasmonate on secondary metabolites of sweet basil (Ocimum basilicum L.). J Agric Food Chem 54:2327–2332
Krithika R, Srivastava P, Rani B, Kolet SP, Chopade M, Soniya M, Thulasiram HV (2015) Characterization of 10-hydroxygeraniol dehydrogenase from Catharanthus roseus reveals cascaded enzymatic activity in iridoid biosynthesis. Sci Rep 5:8258. https://doi.org/10.1038/srep08258
Kumar A, Giridhar P (2015) Salicylic acid and methyljasmonate restore the transcription of caffeine biosynthetic N-methyltransferases from a transcription inhibition noticed during late endosperm maturation in coffee. Plant Gene 4:38–44
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kumar SR, HB S, Nagegowda DA (2018) Terpene moiety enhancement by overexpression of geranyl (geranyl) diphosphate synthase and geraniol synthase elevates monomeric and dimeric monoterpene indole alkaloids in transgenic Catharanthus roseus. Front Plant Sci 9:942
Leonard E, Runguphan W, O’connor S, Prather KJ (2009) Opportunities in metabolic engineering to facilitate scalable alkaloid production. Nat Chem Biol 5:292–300
Li CY, Leopold AL, Sander GW, Shanks JV, Zhao L (2013) Gibson SI. The ORCA2 transcription factor plays a key role in regulation of the terpenoid indole alkaloid pathway. BMC Plant Biol 13:155. https://doi.org/10.1186/1471-2229-13-155
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 25:402–408
Lorence A, Nessler CL (2004) Camptothecin, over four decades of surprising findings. Phytochemistry 65:2735–2749
Magadum S, Banerjee U, Murugan P, Gangapur D, Ravikesavan R (2013) Gene duplication as a major force in evolution. J Genet 92:155–116
Miettinen K, Dong L, Navrot N, Schneider T, Burlat V, Pollier J, Woittiez L, Van Der Krol S, Lugan R, Ilc T, Verpoorte R (2014) The seco-iridoid pathway from Catharanthus roseus. Nat Commun 5:3606
Mizutani M (2012) Impacts of diversification of cytochrome P450 on plant metabolis. Biol Pharm Bull 35:824–832
Moerkercke AV, Steensma P, Schweizer F, Pollier J, Gariboldi I, Payne R, Bossche RV, Miettinen K, Espoz J, Purnama PC, Kellner F (2015) The bHLH transcription factor BIS1 controls the iridoid branch of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Proc Natl Acad Sci 112:8130–8135
Murata J, Roepke J, Gordon H, De Luca V (2008) The leaf epidermome of Catharanthus roseus reveals its biochemical specialization. Plant Cell 20:524–542
Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378
Pan Q, Mustafa NR, Tang K, Choi YH, Verpoorte R (2016) Monoterpenoid indole alkaloids biosynthesis and its regulation in Catharanthus roseus: a literature review from genes to metabolites. Phytochem Rev 15:221–250
Pandith SA, Dhar N, Rana S, Bhat WW, Kushwaha M, Gupta AP, Shah MA, Vishwakarma R, Lattoo SK (2016) Characterization and functional promiscuity of two divergent paralogs of type III plant polyketide synthases from Rheum emodi Wall ex. Meissn. Plant Physiol. https://doi.org/10.1104/pp.16.00003
Pathania S, Bagler G, Ahuja PS (2016) Differential network analysis reveals evolutionary complexity in secondary metabolism of Rauvolfia serpentina over Catharanthus roseus. Front Plant Sci 7:1229
Peebles CA, Hughes EH, Shanks JV, San KY (2009) Transcriptional response of the terpenoid indole alkaloid pathway to the overexpression of ORCA3 along with jasmonic acid elicitation of Catharanthus roseus hairy roots over time. Metab Eng 11:76–86
Pommier Y (2009) DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 109:2894–2902
Pompon D, Louerat B, Bronine A, Urban P (1996) Yeast expression of animal and plant P450s in optimized redox environments. Methods Enzymol 272:51–64
Prall W, Hendy O, Thornton LE (2016) Utility of a phylogenetic perspective in structural analysis of CYP72A enzymes from flowering plants. PLoS One. https://doi.org/10.1371/journal.pone.0163024
Qu Y, Easson ML, Froese J, Simionescu R, Hudlicky T, De Luca V (2015a) Completion of the seven-step pathway from tabersonine to the anticancer drug precursor vindoline and its assembly in yeast. Proc Natl Acad Sci 112:6224–6229
Qu X, Pu X, Chen F, Yang Y, Yang L, Zhang G, Luo Y (2015b) Molecular cloning, heterologous expression, and functional characterization of an NADPH-cytochrome P450 reductase gene from Camptotheca acuminata, a camptothecin-producing plant. PLoS One 10. https://doi.org/10.1371/journal.pone.0135397
Ramesha BT, Amna T, Ravikanth G, Gunaga RP, Vasudeva R, Ganeshaiah KN, Uma Shaanker R, Khajuria RK, Puri SC, Qazi GN (2008) Prospecting for camptothecines from Nothapodytes nimmoniana in the Western Ghats, South India: identification of high-yielding sources of camptothecin and new families of camptothecines. J Chromatogr Sci 46(4):362–368. https://doi.org/10.1093/chromsci/46.4.362
Rather GA, Sharma A, Pandith SA, Kaul V, Nandi U, Misra P, Lattoo SK (2018) De novo transcriptome analyses reveals putative pathway genes involved in biosynthesis and regulation of camptothecin in Nothapodytes nimmoniana (Graham) Mabb. Plant Mol Biol 96:197–215
Sadre R, Magallanes-Lundback M, Pradhan S, Salim V, Mesberg A, Jones AD, DellaPenna D (2016) Metabolite diversity in alkaloid biosynthesis: a multi-lane (diastereomer) highway for camptothecin synthesis in Camptotheca acuminate. Plant Cell 28:1926–1944. https://doi.org/10.1105/tpc.16.00193
Salim V, Wiens B, Masada-Atsumi S, Yu F, De Luca V (2014) 7-deoxyloganetic acid synthase catalyzes a key 3 step oxidation to form 7-deoxyloganetic acid in Catharanthus roseus iridoid biosynthesis. Phytochemistry. 101:23–31
Savatin DV, Gramegna G, Modesti V, Cervone F (2014) Wounding in the plant tissue: the defense of a dangerous passage. Front Plant Sci 5:470. https://doi.org/10.3389/fpls.2014.00470
Shakya P, Marslin G, Siram K, Beerhues L, Franklin G (2019) Elicitation as a tool to improve the profiles of high-value secondary metabolites and pharmacological properties of Hypericum perforatum. J Pharm Pharmacol 71:70–82
Sharma A, Verma P, Mathur A, Mathur AK (2018) Overexpression of tryptophan decarboxylase and strictosidine synthase enhanced terpenoid indole alkaloid pathway activity and antineoplastic vinblastine biosynthesis in Catharanthus roseus. Protoplasma 255(5):1281–1294. https://doi.org/10.1007/s00709-018-1233-1
Shukla AK, Shasany AK, Gupta MM, Khanuja SP (2006) Transcriptome analysis in Catharanthus roseus leaves and roots for comparative terpenoid indole alkaloid profiles. J Exp Bot 57:3921–3932
Singh RS, Kesari R, Kumar U, Jha VK, Kumar A, Kumar T, Pal AK, Singh PK (2018) Candidate genes of flavonoid biosynthesis in Selaginella bryopteris (L.) Baker identified by RNA-Seq. Funct Integr Genomics 18:505–517. https://doi.org/10.1007/s10142-018-0603-2
Sun Y, Luo H, Li Y, Sun C, Song J, Niu Y, Zhu Y, Dong L, Lv A, Tramontano E, Chen S (2011) Pyrosequencing of the Camptotheca acuminata transcriptome reveals putative genes involved in camptothecin biosynthesis and transport. BMC Genomics 12. https://doi.org/10.1186/1471-2164-12-533
Yamazaki M, Mochida K, Asano T, Nakabayashi R, Chiba M, Udomson N, Yamazaki Y, Goodenowe DB, Sankawa U, Yoshida T, Toyoda A (2013) Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant Cell Physiol 54:686–696
Yang L, Ding G, Lin H, Cheng H, Kong Y, Wei Y, Fang X, Liu R, Wang L, Chen X, Yang C (2013) Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS One 8:e80464. https://doi.org/10.1371/journal.pone.0080464
Zhao L, Sander GW, Shanks JV (2013) Perspectives of the metabolic engineering of terpenoid indole alkaloids in Catharanthus roseus hairy roots. Adv Biochem Eng Biotechnol 134:23–54
Acknowledgments
Authors are thankful to Dr. Utpal Nandi at CSIR-IIIM, Jammu for facilitating LC-MS/MS analyses. We are also thankful to Nicolas Navrot, University de Strasbourg, France, for providing pYeDP60 vector and Wat11 strain. GAR is thankful to UGC for providing Senior Research Fellowship. AS thankfully acknowledges the DST-INSPIRE Senior Research Fellowship. This manuscript represents Institutional Communication No. CSIR/IIIM/IPR/0064.
Funding
This work was supported by a financial grant from Council of Scientific and Industrial Research (CSIR)-Indian Institute of Integrative Medicine under Major Lab Project MLP-3012 (WP 7).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: SKL. Performed the experiments: GAR, AS, AK. Analyzed the data: GAR, SKL, VK, PM. Contributed reagents/materials/analysis tools: SKL. Original draft of the manuscript was prepared by GAR. SKL, PM, and VK improved the content and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Handling Editor: Peter Nick
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary file1
Primers used for amplification and expression of NnCYP72A1. (DOCX 17 kb)
Supplementary file 2
Conserved residue prediction for NnCYP72A1 and multiple sequence alignment. (DOCX 324 kb)
Supplementary file 3
Enzyme kinetics of NnCYP72A1. (XLSX 537 kb)
Supplementary file 4
Nucleotide sequences of NnCYP72A1 gene promoter. (DOCX 1111 kb)
Supplementary file 5
Chemical profiles of tissue extracts for detection and quantification of camptothecin. (XLSX 336 kb)
Rights and permissions
About this article

Cite this article
Rather, G.A., Sharma, A., Misra, P. et al. Molecular characterization and overexpression analyses of secologanin synthase to understand the regulation of camptothecin biosynthesis in Nothapodytes nimmoniana (Graham.) Mabb.. Protoplasma 257, 391–405 (2020). https://doi.org/10.1007/s00709-019-01440-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-019-01440-9