
Abstract
In response to environmental light signals, gene expression adjustments play an important role in regulation of photomorphogenesis. LHCB2.4 is among the genes responsive to light signals, and its expression is regulated by redox-regulated members of G-group bZIP transcription factors. The biochemical interrelations of GBF1-interacting protein 1 (GIP1) and the G-group bZIP transcription factors have been investigated. GIP1, previously shown to enhance DNA-binding activities of maize GBF1 and Arabidopsis GBF3, is a plant specific protein that reduces DNA-binding activity of AtbZIP16, AtbZIP68, and AtGBF1 under non-reducing conditions through direct physical interaction shown by the yeast two-hybrid and pull-down assays. Fluorescence microscopy studies using cyan fluorescent protein (CFP)-fusion protein indicate that GIP1 is exclusively localized in the nucleus. Under non- reducing conditions, GIP1 exhibits predominantly high molecular weight forms, whereas it predominates in low molecular weight monomers under reducing conditions. While reduced GIP1 induced formation of DNA-protein complexes of G-group bZIPs, oxidized GIP1 decreased the amount of those complexes and instead induced its chaperone function suggesting functional switching from redox to chaperone activity. Finally analysis of transgenic plants overexpressing GIP1 revealed that GIP1 is a negative co-regulator in red and blue light mediated hypocotyl elongation. By regulating the repression effect by bZIP16 and the activation effect by bZIP68 and GBF1 on LHCB2.4 expression, GIP1 functions to promote hypocotyl elongation during the early stages of Arabidopsis seedling development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Within plant cells, some genes are expressed constitutively, whereas others are expressed in response to specific stimuli (Yamaguchi-Shinozaki and Shinozaki 2006). For the two expression patterns, interaction of transcription factors with cis-acting elements and/or with other transcription factors is required. Among transcription factors, bZIP family comprises one of the more relevant transcription factors involved in the gene regulation. In Arabidopsis, at least 75 predicted bZIP transcription factors have been reported (Jakoby et al. 2002; Deppmann et al. 2004; Vincentz et al. 2003) to be clustered into 10 subgroups A-I and S (Jakoby et al. 2002). The G-group includes AtbZIP16, AtbZIP68, AtGBF1, AtGBF2, and AtGBF3 proteins (Jakoby et al. 2002; Deppmann et al. 2004; Vincentz et al. 2003; Shaikhali et al. 2012). The functional core promoter element specifically bound by plant bZIP transcription factor preferentially contains an ACGT core (Shaikhali et al. 2012; Schindler et al. 1992; Foster et al. 1994; Siberil et al. 2001).
The DNA binding activity of bZIP transcription factors can be altered posttranslationally by 1) heterodimerization, 2) redox-dependent modifications, and 3) protein-protein interaction. AtbZIP16 transcription factor interacts with AtbZIP68 and AtGBF1 in the yeast two hybrid systems (Shaikhali et al. 2012) and heterodimerizes with them upon DNA binding (Shen et al. 2008). In addition, the GBF proteins GBF1, GBF2, and GBF3 heterodimerize with each other suggesting that heterodimer formation increases the diversity of functional Gbox binding proteins (Schindler et al. 1992). Therefore, formation of bZIP homo- or heterodimers offers a tremendous combinatorial flexibility to a regulatory system.
Several transcription factors have been shown to be regulated by redox (Torn et al. 2002; Heine et al. 2004; Shaikhali et al. 2008; Stroher et al. 2009). Among those redox-regulated transcription factors are the typical R2R3 MYB transcriptional regulator P1 protein of maize flavonoid biosynthesis (Heine et al. 2004), a group of plant homeodomain transcription factors (Torn et al. 2002), Rap2.4a, an AP2-domain containing transcription factor involved in the expression of the chloroplast 2CPA (Shaikhali et al. 2008), the stress-associated protein SAP12 (Stroher et al. 2009), the membrane-tethered transcription factor ANAC089 (Klein et al. 2012), and the recently reported members of the group-G bZIP transcription factors, AtbZIP16, AtbZIP68, and AtGBF1 (Shaikhali et al. 2012).
Protein-protein interaction adds another level of control over bZIP transcription factors in which they can be structurally modulated in order to create modified DNA-protein complexes. Pathogen elicitation and salicylic acid-mediated redox changes lead to reduction of NPR1 and translocation of monomers to the nucleus where they interact with several transcription factors such as TGA and modulate transcription of target genes (Tada et al. 2009; Despres et al. 2003). In vitro binding and yeast two-hybrid assays revealed interaction between the GPRI1, GPRI2 transcriptional activators, and the proline-rich region of GBF1, GBF3 that is important for transcriptional modulation of target genes (Tamai et al. 2002). Moreover, GBF1 physically interacts with COP1, suggesting a direct role of COP1 in maintaining the stability of GBF1 protein in light-grown seedlings (Mallappa et al. 2008).
A wide spectrum of light induces photomorphogenesis (Jiao et al. 2007), and therefore, it is not surprising that plants can detect all facets of light, including direction, intensity, duration, and wavelength using three major classes of photoreceptors: the red/far-red light absorbing phytochromes, the blue/UV-A light absorbing cryptochromes and phototropins, and the UV-B sensing UV-B receptor (Chen et al. 2004). Two developmental patterns in the absence or presence of light have been proposed for the Arabidopsis seedling growth (Von Arnim and Deng 1996). Skotomorphogenic (etiolated) growth occurs in darkness and photomorphogenesis (deetiolated) growth occurs in light (Nagatani et al. 1993; Whitelam et al. 1993; Neff et al. 2000). A dramatic morphological change from skotomorphogenic to photomorphogenic growth involves a change in the expression of an estimated one-third of the genes in Arabidopsis (Ma et al. 2001). This massive change in gene expression is mediated by several bZIP transcription factors such as HY5 which has been identified as a positive regulator of photomorphogenesis under various wavelengths of light (Ang and Deng 1994; Pepper and Chory 1997). HYH, a bZIP protein homologue of HY5 acts as positive regulator in blue light-mediated photomorphogenic growth (Holm et al. 2002). Among the group G bZIP subfamily, GBF1 and bZIP16 function as negative regulators of blue and red light-mediated inhibition of hypocotyl elongation and as positive regulators of cotyledon expansion and seed germination, respectively (Mallappa et al. 2006; Hsieh et al. 2012).
In this study, the effect of GIP1 on the DNA-binding affinity of members of the G-group bZIP transcription factors, bZIP16, bZIP68, and GBF1, in a redox-dependent manner was investigated. The physical interaction of GIP1 with these proteins was studied. Moreover, data from transgenic plants indicate that GIP1 protein is a modulator of members of the G-group bZIP transcription and suggest a role in light-regulated photomorphogenesis by co-regulating the expression of LHCB2.4 gene.
Materials and methods
Plant material
Seeds of wild-type Arabidopsis (ecotype Col-0) were stratified at 4 °C for 48 h, and plants were grown on soil at 23 °C (16 h light 100 μmol quanta m−2 s−1) and 18 °C (8 h dark) at 60 % relative humidity.
Total and nuclear protein extraction
Plant tissue of 7-day-old Arabidopsis seedlings was ground to fine powder in liquid nitrogen. Total proteins were extracted in protein extraction buffer (50 mM Tris–HCl, pH 7.5; 150 mM NaCl; 5 mM MgCl2; 10 % glycerol; 0.1 % TritonX-100; 1 mM PMSF and 1 % protease inhibitor cocktail; Sigma). Protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad) according to the manufacturer’s instructions. SDS-loading buffer was added, and the samples were heated for 5 min at 95 °C before loading on 10 % SDS-PAGE.
To isolate nuclear protein extracts (NPE), 5–7-day-old Arabidopsis cell culture line (Col-0) was used. Cells were collected by centrifugation and homogenized in double volume nuclei extraction buffer (NIB; 20 mM Hepes, pH 7.5; 10 mM MgCl2, 10 mM KCL, 1 mM dithiothreitol (DTT) and protease inhibitor cocktail). The cells were lysed using French press at about 5000 psi, and the pellet was collected by centrifugation at 1260 g for 15 min at 4 °C after the cell lysate was filtered through 4-layers of 100-μm filter mesh to remove cell debris. Two washing steps were performed in which the pellet was suspended in equal volume of NIB and centrifuged as above. To extract nuclear proteins, the pellet was suspended in Buffer A-1.2; pH 7.6 (1.2 M KAc, 25 mM Hepes, 10 % glycerol, and 1 mM EDTA), and the mixture was gently sonicated for about 2 min to break the nuclear membranes. Centrifugation at 13,000g for 15 min was performed to collect the supernatant which was subsequently ultra-centrifuged at 37,500g for 20 min to remove DNA. The supernatant was transferred to a fresh tube for protein estimation.
Purification of recombinant proteins
GIP1 (At3g13222) and GBF3 (At2g46270) coding sequences were amplified using NcoI-GIP1-F: 5′- GCTTCCATGGGCAGCAGGATCAGCGGCGAT-3′, Acc65I-GIP1-R: 5′- GCTTGGTACCCTAGTAGTTGTTTCCCAG-3′, NcoI-GBF3-F: 5′-GCTTCCATGGGC GGAAATAGCAGCGAGGA-3′ and Acc65I-GBF3-R: 5′-GCTTGGTACC TCAGCCTGCAGCTACTG-3′ primers, respectively. The PCR products of GIP1and GBF3 were cloned into NcoI/Acc65I sites of pETHIS_1a vector (Department of Chemistry, Umeå University). The resulting plasmids were transformed into E. coli BL21 (DE3) star. To induce the expression of the proteins at 28 °C, IPTG was added at final concentration of 1 mM when OD600 of the cultures was about 0.8 AU and the cultures were grown further for about 16 h. Centrifugation at 6000g, 4 °C for 20 min was performed to harvest the cells which were subsequently lysed by sonication after they were incubated on ice for 30 min in the presence of 1 mg/ml lysozyme. Clear supernatant was collected by centrifugation at 12,000g, 4 °C for 40 min before expressed proteins were affinity purified on Ni2+−NTA agarose resin (Qiagen). bZIP16, bZIP68, and GBF1 were expressed and purified as described previously (Shaikhali et al. 2012).
Electrophoretic mobility shift assays (EMSA)
DNA-protein interactions and electrophoretic mobility shift assays were performed as described previously (Shaikhali et al. 2012). Biotin detection was performed using Chemiluminescent Nucleic Acid Detection Module (Pierce) according to the supplier’s instructions.
Yeast two-hybrid assays
Complementary DNA (cDNA) of GIP1 gene was cloned into pLexA-N vector to generate a fusion protein with LexA DNA binding domain (Bait). AtbZIP16, AtbZIP68, and AtGBF1 full-length cDNAs were cloned into pGAD-HA vector to generate fusions of the prey protein with the GAL4 activation domain (prey). Bait and prey vectors were co-transformed into the NMY51, and transformants were selected on two selective media lacking tryptophan, leucine, and histidine (SD/-Trp-Leu-HIS). The competitive inhibitor, 3-aminotriazole (3AT), of the leaky expression of the HIS gene was used when needed. The yeast two-hybrid assay was performed with the DUALhybrid system (Biotech) according to the manufacturer’s instructions. Primer sequences used in the bait construct for GIP1 were EcoRI-GIP1-F: 5′-AAAAGAATTCATGAGCAGGATCAGCGGCG-3′ and KpnI-GIP1-R: 5′-AAAAGGTACCCTAGTAGTTGTTTCCCAGA-3′. The primers used to generate cDNAs for prey cloning are the following: BamHI-bZIP16-F 5′-AAAAGGATCCATGGCTAGCAATGAGATGG-3′ and XhoI-bZIP16-R 5′-AAAACTCGAG ACTCGTTGAGTCTTTGTATGAAT-3 for AtbZIP16, BamHI-bZIP68-F: 5′-AAAAGGATCCATGGGTAGCAGTGAGA-3′ and XhoI-bZIP68-R: 5′-AAAACTCGAGCTACGCAACATCCTGA-3′ for AtbZIP68 and BamHI-GBF1-F: 5′-AAAAGGATCCATGGGAACGAGCGAAGAC-3′ and XhoI-GBF1-R: 5′-AAAACTCGAGTTAATTTGTTCCTTCACC-3′ for AtGBF1. For β-galactosidase liquid assay, fresh overnight cultures of NMY51 harboring the desired constructs were diluted in YPDA to OD600∼0.1 before cells were grown further for 3–5 h (log phase ∼0.5–0.8). Before cells were harvested, the exact OD600 was recorded, individual cultures were divided into three tubes, and cells were collected by centrifugation. Cells were washed with 1.5 ml Z buffer; pH 7.0 (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4). Subsequently, cells were suspended in 0.3 ml Z buffer, and 100 μl aliquots were removed and transferred into fresh tubes. After three freezing and de-freezing series, 0.7 ml of Z buffer containing 0.27 % 2-mercaptoethanol were added. Time was recorded before addition of 160 μl of 4 mg/ml o-nitrophenyl β-d-galactopyranoside (ONPG; Duchefa). Reactions and blank were incubated at 30 °C and 0.4 ml of 1 M Na2CO3 was added when a yellow color was developed. After centrifugation for 10 min, the supernatant was transferred into fresh cuvettes and A420 was measured. β-galactosidase units were calculated according to the following equation: β-galactosidase unit = 1000 × OD420/t × v × OD600 where (t) is the elapsed time of incubation and (v) is the volume of 0.1 ml multiplied by concentration factor.
Pull-down assays
Using His-tagged bZIP16, bZIP68, and GBF1 proteins and NPEs from Arabidopsis cell culture, pull-down assays were performed with PierceTM Pull-Down PolyHis Protein:Protein Interaction Kit (Thermo Scientific) according to the manufacturer’s instructions.
Subcellular localization
Full-length coding sequence of GIP1 lacking the stop codon was amplified by PCR. The amplified PCR product was cloned into BamHI/KpnI and fused to the N terminus of CFP in the 35S-CFP vector to generate the construct GIP1-CFP. GIP1-CFP and ABI5-YFP were transfected and co-expressed transiently in protoplasts and incubated for 16 h in the dark. SP2 confocal laser scanning microscope (Leica) was used to visualize the fluorescence. All micrographs were taken with ×63 water immersion objective with a numerical aperture of 0.75. Images were taken at 433 and 514 nm specific for CFP and YFP, respectively.
Blue native and sodium dodecyl sulfate polyacrylamide gel electrophoresis (BN- and SDS-PAGE)
BN-PAGE was used as described in Eubel et al. (2005) with some modifications. Recombinant GIP1 protein with or without DTT was incubated in blue native solubilization buffer (10 % glycerol, 4 % digitonin, 1 % β-dodecylmaltoside, 150 mM KAc and 30 mM Hepes, pH 7.4) for 10 min on ice before it was mixed with 5× native sample buffer (62.5 mM Tris-HCl, pH 6.8, 40 % glycerol and 0.01 % Bromophenol Blue; BioRad). The protein samples were loaded on 4–15 % Mini-PROTEAN® TGX™ Precast Gel (BioRad) and running was performed using 1× Tris/glycine buffer. For two-dimensional (2D) native SDS-PAGE, single-lane first-dimension (BN-PAGE) strips were excised from native gels, followed by incubation in reducing 1× running buffer (25 mM Tris, 192 mM glycine, 0.1 % SDS) supplemented with 50 mM DTT for 30 min. Equilibrated gel strips were applied to a classical SDS-PAGE gels and running was performed using 1× running buffer to separate the complexes (oligomers) according to their molecular weight. The gel was blotted on PVDF membrane, blocked with powder milk in 1× TBST, incubated with anti-HIS antibody and detected with ECL plus Western blotting detection system (GE healthcare) according to the supplier’s protocol.
Rhodanese refolding assay
In order to prepare denatured rhodanese, lyophilized rhodanese was solubilized to 25–40 μM in 6 M guanidine-HCl, 10 mM Tris; pH 7.4. To initiate folding, diluting denatured rhodanese to a final concentration of 100 nM in buffer B (25 mM Tris; pH 7.4, 20 mM Na2SO3, 12 mM MgCl2, and 5 mM KCl) supplemented with/without GIP1 as indicated in the legend to Fig. 7. The reactions were completed after 60 min incubation at 25 °C and then quenched by addition of 30 mM EDTA. Finally, rhodanese enzyme activity was determined as described previously (Tandon and Horowitz 1989).
Hypocotyl measurement and gene expression
For hypocotyl measurement and LHCB2.4 gene expression, Arabidopsis seedlings were grown on Murashige-Skoog plates without sucrose. After cold stratification for 24 h at 4 °C in darkness, the plates were placed in 150 μmol m−2 s−1 constant white light for 12 h to induce germination, and then dark-adapted for 24 h prior to 5-days growth in either 5 μE continuous red light or 20 μE continuous blue light. Hypocotyl length was measured by use of the software ImageJ (http://rsbweb.nih.gov/ij/). The means and standard deviations were calculated from 60 to 80 seedlings. Total RNA isolation, cDNA synthesis, and quantitative PCR were as described previously (Shaikhali et al. 2012).
Results
The DNA-binding activity of AtbZIP16, AtbZIP68, and AtGBF1 is modulated by GIP1
Accessory proteins have been reported to modulate bZIP DNA-binding activity (Despres et al. 2003; Virbasius et al. 1999; Sehnke et al. 2005). Using Arabidopsis thaliana cDNA library, GIP1 was isolated in a yeast two-hybrid screen for proteins interacting with ZmGBF1. GIP1 interacts with ZmGBF1 and enhances its DNA-binding activity (Sehnke et al. 2005). While Zea mays GBF1 transcription factor is involved in Adh regulation, Arabidopsis GBF1, in addition to bZIP16 and bZIP68, regulates LHCB2.4 expression in a redox-dependent manner during light-regulated photomorphogenesis (Shaikhali et al. 2012). Like ZmGBF1, GIP1 enhanced DNA-binding activity of AtGBF3 (Fig. 1a; Sehnke et al. 2005). The bZIP domain shares about 60 % identities among ZmGBF1, AtGBF3, and AtGBF1 (Fig. S1A). Whereas, ZmGBF1 shares 38.5 and 34.5 % overall identity with AtGBF3 and AtGBF1, respectively, AtGBF3 and AtGBF1 share 31 % overall homology (Fig. S1A). There is higher sequence similarity between ZmGBF1 and AtGBF3 (Fig. S1B).

GIP1 protein modulates DNA-binding activities of AtbZIP16, AtbZIP68 and AtGBF1. a EMSA was performed with binding reactions containing biotin-labeled DNA probe (G-box) and was incubated alone (lane 1), with GIP1 (lane 2) and with GBF3 and GIP1 (lane 3). b EMSAs binding reactions contained biotin-labeled probe (G-box) incubated without protein (lane 1), or with GIP1 (lane 2), bZIP16 (lane 3), bZIP16 and GIP1 (lane 4), bZIP68 (lane 5), bZIP68 and GIP1 (lane 6), GBF1 (lane 7), GBF1 and GIP1 (lane 8). c To validate the effect of GIP1, E. coli protein extract and BSA were used. EMSAs were performed and the binding reactions contained biotin-labeled DNA probe (G-box) and bZIP16, bZIP68 or GBF1 (lane 1 in each panel). E. coli protein extract and BSA were added to the DNA-protein complexes (lane 2 and 3 in each panel). Biotin-labeled probes were detected with chemiluminescent nucleic acid detection module and positions of free DNA and protein-DNA complexes are indicated by an arrow. Experiments were performed at least three times and showed similar results in each case
It was investigated whether DNA-binding activity of members of G-group transcription factors can be catalyzed by the nuclear protein GIP1. AtbZIP16, AtbZIP68, and AtGBF1 proteins were expressed as HIS-tagged recombinant proteins as described previously (Shaikhali et al. 2012). Moreover, GIP1 was also expressed as HIS-tagged recombinant protein in E. coli. Equal amounts of GIP1 protein were added to binding reactions containing AtbZIP16, AtbZIP68, or AtGBF1 recombinant proteins and Gboxcis (5′-TCAACTGACACGTGGCATAAC-3′) as a probe. While GIP1 enhanced DNA-binding activity of AtGBF3 (Fig. 1a, lane 3; Sehnke et al. 2005), it reduced DNA-binding activity of AtbZIP16, AtbZIP68, and AtGBF1 under non-reducing conditions (Fig. 1b; lanes 4, 6, 8). By itself, GIP1 did not interact with the Gbox of the Adh promoter (Sehnke et al. 2005) or with the Gbox DNA probe of LHCB2.4 promoter (Fig. 1b; lane 2), suggesting that GIP1 is not a DNA-binding protein. Moreover, the migration mobility of each respective protein-DNA complex was not changed or qualitatively altered (Fig. 1b) suggesting that GIP1 does not participate in the DNA-protein complex.
To prove that the effect on the DNA-protein complexes of AtbZIP16, AtbZIP68, and AtGBF1 was due to GIP1, either BSA or E. coli cell extract were added to the binding reactions. Both BSA and E. coli cell extract were not effective and did not change the migration mobility or pattern (Fig. 1c; lane 2 and 3). These results indicate that there is a requirement for a specific accessory factor rather than simply a proteinaceous environment for DNA binding by those G-group bZIP transcription factors. The reduction in the DNA-protein complexes upon addition of recombinant GIP1 (Fig. 1b) indicates that probably, GIP1 is responsible for such an effect.
Physical interaction of GIP1 with bZIP16, bZIP68, and GBF1 proteins
The activities of many regulatory transcription factors involve interactions with other proteins (Chi et al. 2013). To investigate whether the effects generated by GIP1 on the DNA-protein complex affinities was due to direct physical interaction between GIP and the G-group bZIP transcription factors, yeast two-hybrid assays were performed. GIP1 was expressed as fusion to the GAL4-DNA binding domain in pLexAN vector (BD-GIP1) and bZIP16, bZIP68, and GBF1 as fusions to the GAL4 activation domain in pGADHA vector (AD-bZIP16, AD-bZIP68, and AD-GBF1), respectively. Combinations of bait and prey were introduced into NMY51 yeast strain containing HIS and/or LacZ genes under the control of GAL4 binding sites (Fig. 2). Selective media lacking the nutritional selective markers SD-Trp, Leu or SD- Trp, Leu, HIS was used to grow the transformants. 3-AT was used to eliminate the leaky expression of the HIS gene, due to self-activating capacity of GIP1. BD-GIP1 bait was able to interact with the preys AD-bZIP16, AD-bZIP68, and AD-GBF1; however, BD-GIP1 did not interact with the empty AD vector (Fig. 2a). To further substantiate observed interaction between GIP1 and these proteins, chimeric fusion proteins of GAL4 activation domain AD-bZIP16, AD-bZIP68, or AD-GBF1 activated the transcription of LacZ reporter gene significantly in the presence of BD-GIP1. When GIP1 was expressed alone, in the presence of empty AD vector, no significant increase in the expression of LacZ reporter was observed (Fig. 2b). Taken together, these data suggest direct physical protein-protein interaction between GIP1 and these proteins in vivo in the yeast cells.

Physical interaction of GIP1 with bZIP16, bZIP68, and GBF1. Full-length coding sequence of GIP1 was fused to Gal4-binding domain in pLexA-N vector to generate the bait BD-GIP1. bZIP16, bZIP68, or GBF1 were fused to the Gal4 activation domain in pGADHA vector to produce the prey clones, respectively. NMY51 yeast strain was co-transformed with BD-GIP1 bait and AD-bZIP16, AD-bZIP68, AD-GBF1 prey constructs. a Combinations of BD-GIP1 and pGADHA empty prey vector represent a negative control. BD-GIP1/AD-bZIP16, BD-GIP1/AD-bZIP68, and BD-GIP1/AD-GBF1 are interacting targets. Interaction is indicated by the growth of yeast cells on -HIS selective media in the presence of 10 mM 3-AT and the two rows in each panel are two replicates from independent co-transformations. b Activation of lacZ reporter was measured by quantitative β-galactosidase (β-gal) activity assays. β-galactosidase activity was measured for each transformant. Six measurements obtained from two independent yeast clones are given. Error bars indicate SE (n = 6) according to the student’s t test and the asterisks (*) indicate significant difference P < 0.001. The enzyme activity of the empty vector was defined as 1.0. c In vitro pull-down assay of His −bZIP16, −bZIP68, and −GBF1 purified proteins and nuclear protein extracts (NPEs) of Arabidopsis cell culture. His-tagged proteins were incubated with the NPEs and pulled-down proteins were purified with Ni-NTA beads. Ni-NTA beads alone were used as a negative control. The eluted samples were separated by 10%SDS-PAGE, blotted and probed with anti-GIP1 antibody. Lanes: 1 bZIP16 bait control, 2 bZIP68 bait control, 3 GBF1 bait control, 4 proteins pulled down from NPEs and Ni-NTA beads alone 5–7, proteins pulled down from NPEs by His bZIP16, −bZIP68 and GBF1. Molecular mass marker (kDa) is shown on the left and the GIP1 protein signal is indicated by an arrow
To verify the interaction between GIP1 and the three transcription factors in vitro, His pull-down assays were performed. The purified His-tagged bZIP16, bZIP68, and GBF1 proteins were incubated with the nuclear protein extracts (NPEs) from Arabidopsis cell culture. After incubation, His-bZIP16, −bZIP68 and −GBF1 and bound protein(s) were purified by affinity chromatography. Samples were separated by SDS-PAGE and probed with anti-GIP1 antiserum. A negative control using the Ni-NTA beads was set up to exclude the possibility that NPEs interact directly with the NTA resin. As shown in (Fig. 2c; lanes 5–7), ∼65 KDa band corresponding to the size of GIP1 protein was pulled down and captured by the purified His-tagged proteins. In the negative control, nothing was detected in the Ni-NTA pull-down products using NPEs lacking the His-tagged proteins (Fig. 2; lane 3). Therefore, the pull-down assay with the His-tag proteins confirmed the interaction between GIP1 and bZIP16, bZIP68 and GBF1 transcription factors in vitro.
Subcellular localization and secondary structure of GIP1
Analysis of secondary structure of GIP1 protein did not reveal any amino acid sequence indicating a putative nuclear localisation signal (NLS); however, it was previously shown that GIP1 is localized in the nuclei of Arabidopsis after analysis of immunoblots for cytoplasmic and nuclear A. thaliana tissue fractions (Sehnke et al. 2005). To demonstrate the reliability of localization in the fractions prepared from Arabidopsis tissue (Sehnke et al. 2005), nuclear protein extracts (NPE) were purified from nuclei of Arabidopsis cell cultures. In the Western blot analysis using anti GIP1 antibody (Sehnke et al. 2005), a protein of the desired (∼65 KDa) corresponding size to GIP1 protein was detected in the nuclear fraction (Fig. 3a, right) in agreement with the localization results revealed with Arabidopsis tissue (Sehnke et al. 2005). In total protein extract (TPE) prepared from the cell culture, a protein with a similar size to that of the nuclear fraction was detected (Fig. 3a, left).

Subcellular localization of GIP1. a Immunoblot analysis of total protein extract (TPE) and nuclear protein extract (NPE) fractions from Arabidopsis cell culture. The membranes were probed with anti-GIP1 (top) or anti-histonH1 (middle) and anti- HSP70 (bottom) antibodies, respectively. b Nuclear localization of GIP1. ABI5-YFP and GIP1-CFP were co-transfected into Arabidopsis thaliana mesophyll protoplasts. Confocal images are 1 bright field transmission, 2 chlorophyll autofluorescence. 3 ABI5-YFP fluorescence, 4 GIP1-CFP fluorescence, 5 the merged image of 1–4. Bar length = 20 μm. The arrows indicate a successfully transformed representative protoplast
The subcellular localization of GIP1 protein was investigated further in transiently transfected protoplasts from Arabidopsis using confocal laser scanning microscopy (CLSM). The GIP1-CFP fusion protein was exclusively localized to the nucleus (Fig. 3b). ABI5-YFP fusion protein was used as a nuclear marker (Shaikhali et al. 2012) that exclusively co-localized with GIP1 in the nucleus (Fig. 3b). The fluorescence distribution of GIP1-CFP in the nucleus was consistent with expected nuclear localization of this protein and confirms that GIP1 is likely localized to the nucleus.
GIP1 was subjected to secondary structure analysis to search for predicted motifs which could be linked to functionality. Apart from DUF1296, a conserved region of unknown function of approximately 60 amino acid residues long within a number of plant proteins, no similarity to known motifs was revealed (Fig. 4a). Since a protein’s secondary structure can be predicted from its sequence based on the intrinsic propensities of its individual amino acids to adopt helical, sheet or turn confirmation, secondary structure prediction performed using YASPIN secondary structure prediction (http://www.ibi.vu.nl/programs/yaspinwww/) resulted in the presence of several α-helices, few β-sheets and lots of coils (Fig. 4a). Moreover, analysis of GIP1 protein sequence for possible posttranslational modifications revealed several CK2 and PKC phosphorylation sites, several ASN glycosylation sites, several myrostylation sites, and one amidation site. Interestingly, GIP1 contains 4 Cys amino acid residues which might be susceptible to oxidation and formation of disulfide bond(s) which serve an important structural role in many proteins.

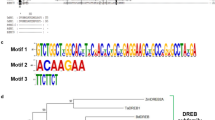
Secondary structure analysis and phylogenetic tree of GIP1 protein. a Amino acid sequence of GIP1 with its corresponding secondary structure α-helical parts labeled with (α), extended strand (β), and random coil (c). The asterisks (*) indicate the plant specific DUF1296 domain with unknown function, and the Cys residues are indicated by boxes. b Based on amino acid sequence alignment of GIP1 paralogs and orthologs, the phylogenetic tree was constructed using Phylogeny.fr (http://www.phylogeny.fr). The tree indicates the GIP1 homologues of the different plant species
A blast search in TAIR (http://www.arabidopsis.org/) for paralogs of GIP1 in A. thaliana revealed a kinase-related protein of unknown function (At3g07660). This protein shares 41 % identity with GIP1 and the highest conservation within the N-terminal region where DUF1296 region is located (Fig. 4b). Like GIP1, known motifs which could be linked to functionality were not predicted.
Database searches, using NCBI Blast (http://www.ncbi.nlm.nih.gov/), revealed only plant-specific orthologs of GIP1. The identified orthologs are predicted and/or hypothetical proteins of unknown function that were clustered by constructing a phylogenetic tree (Fig. 4b). Interestingly, identified proteins of lower plants Chlorella variabilis (single cell green algae), Physcomitrella patens subsp. patens (moss) and Selaginella moellendorffii (spikemoss lycophyte) were clustered together quite distantly from Arabidopsis GIP1(Fig. 4b). However, orthologs of the monocots Z. mays, Sorghum bicolor, Orzya sativa subsp. japonica and indica made another cluster closer to the Arabidopsis GIP1 (Fig. 4b). Finally, the orthologs of the dicots Populus trichocarpa, Ricinus communis, and Arabidopsis lyrata made a distinct cluster with A. thaliana GIP1 (Fig. 4b).
Redox-regulation of GIP1 protein
GIP1 protein contains four cysteine residues at positions 45, 234, 288, and 362 (Fig. 4b). The fact that this protein contains Cys residues initially suggested that it could be involved in the formation of intra- or intermolecular disulfide bonds. Analyzing GIP1 protein for disulfide bond predictions by DiANNA (http://clavius.bc.edu/~clotelab/DiANNA/), revealed several putative disulfide bonds between Cys45 and Cys234, Cys288 and Cys362 and Cys45 with Cys362. Denaturing polyacrylamide gels (SDS-PAGE) with GIP1 protein subjected to reducing or oxidizing conditions were performed. Under non-reducing conditions, GIP1 formed several species with apparent molecular masses corresponding to monomeric, dimeric, and oligomeric forms (Fig. 5a; lane 1). However, the predominant form observed for GIP1 in non-reducing conditions corresponded to apparent molecular masses of oligomers. The amounts of GIP1 oligomers/dimers were significantly reduced after treatment with increasing concentrations of DTT (Fig. 5a; lanes 2–4). On the other hand, treatment with increasing H2O2 concentrations did not modify the proportion observed under non-reducing conditions suggesting that GIP1 cannot be further oxidized (data not shown).

GIP1 protein analysis under reducing and oxidizing conditions. a Recombinant GIP1 was incubated in the presence of varying concentrations of DTT (0–10 mM). Protein samples were electrophoresed onto 10 % SDS-PAGE and revealed by Coomassie blue staining (top panel) or Western-blot analysis using anti-HIS antibody (lower panel). Protein signals are indicated by arrows. b BN-PAGE analysis of the recombinant GIP1 under non-reducing (0 mM DTT; top) and reducing (10 mM DTT; bottom). Complexes in first dimension were separated on 4–16 % BN-PAGE. Molecular masses were estimated from the marker proteins as shown at the top of the panel. Second dimension was separated on 12 % SDS-PAGE. Positions of molecular weight markers are indicated at the left. The proteins were analyzed after transfer onto nitrocellulose membrane and detected with anti-HIS antibody. c Total protein (100 μg) extracted from 7-day-old wild-type Columbia (Col-0) plants treated with 0 to 2 mM H2O2, either left in their native state (lanes 1–3) or reduced with 10 mM DTT (lanes 4–6), was subjected to 10%SDS-PAGE and analyzed by immunoblot using a anti-HIS antibody. Protein signals are indicated by arrows. Experiments were performed at least three times and showed similar results in each case
To further investigate higher-order GIP1 oligomers, BN/SDS-PAGE was applied. To allow direct comparison of two different treatments (0 and 10 mM DTT) of GIP1 protein, both were subjected to first and second dimension by applying the samples to the same gel in parallel. After BN/SDS-PAGE of recombinant GIP1 under non-reducing conditions (0 mM DTT), anti-HIS immune-blot detection indeed confirmed the separation into higher amounts of distinct complexes of various sizes (Fig. 5b, top). Based on comparison with molecular weight markers, the size of recombinant GIP1 reached from about 73 kDa, in the high percentage area of the gel, up to very high-order oligomers in the MDa range, in the low percentage area of the gel (Fig. 5b, top). The lowest apparent molecular mass likely represents the monomer of the GIP1 complex, albeit migrating at a somewhat higher molecular mass than calculated from the primary sequence with the HIS tag (∼69 KDa). Several distinct signals are detectable at much higher molecular masses, indicating existence of defined homo-oligomers of GIP1 formed probably through disulfide bonds (Fig. 5b, top). Furthermore, GIP1 treatment with DTT prior to the BN-PAGE first dimension was investigated. In the high percentage area of the gel, similar to the non-reducing conditions, the lowest apparent molecular mass likely represents the monomer of the GIP1 (Fig. 5b, bottom). Moreover, lower amounts of distinct signals were also detectable at higher molecular masses indicating the existence of GIP1 oligomers; however, these oligomers are probably formed by other means such as hydrophobic interactions (Fig. 5b, bottom). Taken together, biochemical analysis through BN-PAGE/2D thus demonstrates that GIP1 predominantly migrates in the form of assembled homo-oligomers.
To determine whether the cysteines in GIP1 affect the conformation of the protein in response to redox stress in vivo, protein extractions in the absence of DTT-reducing agent were performed. As a control, DTT (50 mM) was later added to aliquots of the same extracts. Protein extracts from 7-day-old wild-type Columbia (Col-0) seedlings treated with/out H2O2 were subjected to SDS-PAGE, followed by immunoblot analysis. In the absence of H2O2 (control conditions), several bands corresponding to the monomer, dimer, and oligomer of GIP1 were detected (Fig. 5c; lane 1). One millimolar H2O2 treatment showed almost a similar pattern to that of the control conditions (Fig. 5c; lane 2). In response to 2 mM H2O2 treatment, the concentration of monomers, dimers, and oligomers were significantly reduced, suggesting formation of high molecular mass oligomers that were beyond the migration capacity of the gel (Fig. 5c; lane 3). The addition of DTT to the extracts eliminated the oligomers and reduced all the GIP1 protein to the monomeric form (Fig. 5c; lanes 4–6). The reduced monomeric form of GIP1 observed in the immunoblot analysis probably represents an in vivo conformation of the protein. However, to rule out the possibility that the high MW protein complex detected in the immunoblot analysis could be a product of nonspecific crosslinking caused by disulfide bond formation during sample preparation, plant tissue was homogenized and denatured the protein in the presence of the alkylating agents, 2 mM iodoacetamide (IOD) or N-ethylmaleimide (NEM), which block free thiol groups from forming nonspecific disulfide bonds while keeping existing disulfide bonds intact. Treatment with IOD or NEM did not affect the formation of the high MW protein complex (data not shown), confirming that high molecular mass oligomers existed in intact plant cells prior to protein extraction.
Redox-regulated GIP1 regulates DNA-binding activity of members of G-group bZIP transcription factors
The DNA-binding activity of bZIP16, bZIP68, and GBF1 transcription factors was shown to be directly regulated by the changes in their redox state (Shaikhali et al. 2012). Under reducing conditions, binding activity of these proteins was greatly enhanced suggesting that cysteines in the reduced state are required for more efficient binding (Shaikhali et al. 2012). In order to identify the individual role of GIP1 in redox regulation of DNA-binding activity of these proteins, GIP1 was added to the binding reactions as untreated control, reduced, or oxidized. As shown in Fig. 6, presence of untreated GIP1 significantly reduced DNA-binding activity of bZIP16, bZIP68, and GBF1 as determined by EMSA (Fig. 6a–c; lane 2). However, oxidized GIP1 did not influence DNA-binding activity of bZIP16, bZIP68, and GBF1 (Fig. 6a–c; lane 4). This is feasible since untreated and H2O2-oxidized GIP1 showed almost the same quaternary structures (Fig. 5). In contrast, presence of reduced GIP1 revealed enhanced DNA-binding activity of bZIP16, bZIP68, and GBF1 (Fig. 6a–c; lane 3). These results greatly support importance of the disulfide bonds in redox-regulated GIP1 to regulate activities of members of G-group bZIP transcription factors.

Oxidation-reduction of GIP1 differentially affects DNA-binding activities of AtbZIP16, AtbZIP68, and AtGBF1. The effect of untreated, reduced, and oxidized GIP1 on DNA-binding activity of a AtbZIP16, b AtbZIP68, and c AtGBF1 was tested by EMSA. EMSA was performed using biotin-labeled complementary synthetic oligonucleotides representing the G-box element. Biotin-labeled probes were detected with a chemiluminescent nucleic acid detection module and positions of free DNA, and protein-DNA complexes are indicated by arrows. Experiments were performed at least three times and showed similar results in each case
Chaperone activity of GIP1
GIP1 was previously shown to function as molecular chaperone under non-reducing conditions (Sehnke et al. 2005). To explore the relationship between the monomeric/oligomeric status of GIP1 and its function as chaperone, reduced and oxidized GIP1 proteins were examined for general chaperone activity on denatured enzyme rhodanese. Oxidized GIP1 exhibited fourfold higher chaperone activity than that of the negative control (Fig. 7). In contrast, reduced GIP1 displayed almost similar activity measurements to those of the negative control (Fig. 7). From these results, it is concluded that the chaperone function of GIP1 is likely linked to its oligomerization status.

Chaperone activity of GIP1 enhances heat shock recovery in Arabidopsis. a, The ability of reduced (filled square) and oxidized (filled triangle) GIP1 protein to refold urea-denatured rhodanese was compared to the buffer (filled diamond) used in the reactions; 0.1 mg/ml of reduced GIP1, oxidized GIP1, or equivalent buffer was incubated with rhodanese in the absence of ATP. Experiments were performed at least three times and showed similar results in each case. b Comparison of heat shock tolerance between WT and transgenic plants overexpressing GIP1. Plants were grown for 4-weeks at 22 °C (top) then exposed for heat shock by incubation at 45 °C for 5 days. Recovery stage was performed at 22 °C for 5 days (bottom). A scheme of heat shock treatment and recovery of the Arabidopsis seedlings is depicted (middle)
We further investigated the physiological consequences of heat-shock in Arabidopsis WT and transgenic lines overexpressing GIP1 (35S:GIP1). The GIP1 overexpression lines (Lee et al. 2014) showed no phenotypic difference from the WT under normal growing conditions (22 °C; Fig. 7b, top). WT plants and the GIP1 overexpression lines were damaged by the heat stress incurred at 45 °C for 5 days. However, when the heat-stressed plants were returned to their optimal temperature, the transgenic lines overexpressing GIP1 rapidly recovered during the post-stress recovery period (Fig. 7b, bottom). The observation is in good agreement with the results of Arabidopsis stromal HSP70 (Su and Li 2008). In contrast, the WT plants were unable to recover from the damage caused by heat shock (Fig. 7b, bottom). These results suggest that the chaperone function of GIP1 plays a major role in the protection of Arabidopsis from heat shock during the heat stress and/or the recovery stage.
GIP1 overexpression display red light- (RL) and blue light- (BL) specific regulation of hypocotyl elongation and LHCB2.4 gene expression
Because bZIP16 and GBF1 function as negative regulators in RL and BL-mediated hypocotyl elongation, the question was whether their interacting partner GIP1 displays a similar effect. Several independent transgenic lines overexpressing GIP1 under the control of CaMV 35S promoter (Lee et al. 2014) were used. Examination of the hypocotyl length (photomorphogenic response) revealed that the 35S:GIP1 transgenic lines displayed significant reduction of inhibition in hypocotyl elongation in response to R and BL, respectively (Fig. 8a, c). GIP1 transcript levels showed dramatically elevated levels of expression of this gene in overexpression lines relative to wild-type background (Lee et al. 2014). These results indicate that the altered phenotypes of the overexpression lines observed were likely to be caused due to the elevated levels of GIP1. Taken together, these results indicate a biological significance and a functional involvement in planta and suggest that GIP1 is a transcriptional co-regulator of photomorphogenic growth that promotes hypocotyl elongation during early seedling development in Arabidopsis.

GIP1 regulates RL- and BL-mediated photomorphogenesis in Arabidopsis. Hypocotyl lengths of 5-day-old seedlings of WT and transgenic lines overexpressing GIP1 (35S:GIP1) grown in constant a RL (5 μmol m−2 s−1) or c BL (20 μmol m−2 s−1). Asterisk indicates hypocotyl length of 35S:GIP1 lines are significantly different from that of WT (*P, 0.05; **P, 0.01; and ***P, 0.001; Student’s t test; n = 60 to 80). Q-PCR analysis of LHCB2.4 transcript levels in response to b RL (5 μmol m−2 s−1) or d BL (20 μmol m−2 s−1) in 5-day-old seedlings of WT and transgenic lines overexpressing GIP1 (35S:GIP1). The gene expression was normalized to the expression level of At4g36800, which encodes ubiquitin-protein ligase-like protein. The mean ± SD of at least three biological replicates is shown. The expression was significantly different in 35S:GIP1 transgenic lines compared to WT as demonstrated by Student’s t test (*P, 0.05; **P, 0.01; and ***P, 0.001)
The upregulation of light-inducible genes such as CAB/LHCB2.4/RCBS is one of the important phenomena in photomorphogenic growth. Because bZIP16 is a transcriptional repressor and bZIP68 and GBF1 are transcriptional activators in light-regulated expression of LHCB2.4 (Shaikhali et al. 2012), the role of their interacting partner, GIP1 in light-regulated gene expression of LHCB2.4 in planta was investigated. Moreover, bZIP16 regulates the growth of hypocotyls in response to RL (Hsieh et al. 2012) and bZIP68 and GBF1 regulate the growth of hypocotyls in response to BL (Shaikhali unpublished; Mallappa et al. 2006). For this purpose, expression of LHCB2.4 gene was investigated in 5-day-old seedlings of 35S:GIP1 transgenic seedling lines grown in constant RL or BL and compared with WT. The expression level of LHCB2.4 was significantly lower in the 35S:GIP1 transgenic lines compared with WT in response to RL (Fig. 8b), suggesting that GIP1 acts as a co-repressor of LHCB2.4. In contrast and similarly to bZIP68 and GBF1, the 35S:GIP1 transgenic lines displayed significantly higher expression level of LHCB2.4 compared to WT in response to BL (Fig. 8d), suggesting that GIP1 acts as a co-activator of LHCB2.4. Taken together, these results suggest that GIP1 acts as a transcriptional co-regulator of LHCB2.4 gene expression in RL- and BL-mediated inhibition of hypocotyl elongation and provide biological relevance for the interaction with bZIP16, bZIP68, and GBF1 transcription factors.
Discussion
Transcription factors play crucial roles in almost all biological processes and they are important in the regulation of cell activities (Liu et al. 1999). The aim was to identify protein(s) which might contribute to/modulate DNA-binding activity of members of the G-group bZIP transcription factors. Here, it is shown that the regulatory nuclear factor GIP1 does not interact with DNA in absence or presence of bZIP proteins and it is not a component of protein-DNA complex (Fig. 1a). However, this accessory factor negatively regulates affinity interaction of AtbZIP16, AtbZIP68, and AtGBF1 transcription factors with DNA under non-reducing conditions. That suggests a potential regulatory role of GIP1 factor that involves posttranslational modification of DNA-binding activity of bZIP transcription factors (Figs. 1 and 2). GIP1 interacted with these proteins in vivo in the yeast two-hybrid system and in vitro by pull-down assays (Fig. 2). Since interacting proteins must be simultaneously present in the cell/tissue, their lifetimes between synthesis and degradation should overlap. In many cases, co-expression of genes encoding interacting proteins indicates that they are members of the same pathway. Transcript levels of GIP1, bZIP16, bZIP68, and GBF1 genes were closely co-expressed during different stages of plant development (Fig. S2A). In mammals and plants, accessory proteins have been shown to positively and negatively modulate DNA-binding activity of transcription factors (Despres et al. 2003; Ron and Habener 1992). The regulatory protein NPR1 has been shown to positively regulate DNA-binding activity of TGA bZIP transcription factors (Despres et al. 2003). CHOP10, an animal protein, has been identified as a negative regulator of C/EBP bZIP transcription factor (Ron and Habener 1992). Moreover, GPRI1 and GPRI2 proteins were shown to interact with the proline-rich region of GBF1 and GBF3 transcription factors to modulate the transcriptional level of corresponding genes (Tamai et al. 2002).
Since its isolation and initial characterization in 2005 (Sehnke et al. 2005), no biochemical, genetic, or physiological data have been released on GIP1 until only recently when a report by Lee et al. (2014) suggested that GIP1 might act as co-activator to enhance transcriptional activity of the LATERAL ORGAN BOUNDARIES DOMAIN 18 (LBD18) transcription factor (Lee et al. 2014). GIP1 is a plant-specific protein with one paralog, a kinase-related protein of unknown function (GIP1L; At1g55820), from A. thaliana (Fig. 4b; Lee et al. 2014). Unlike GIP1, GIP1L protein contains a transcription activating domain and cannot interact with LBD18 in yeast (Lee et al. 2014). GIP1 and GIP1L transcripts were detected in all tissues examined such as seedlings, root, leaf, stem, and flower (Lee et al. 2014). Orthologs of GIP1 were also identified from different plant species (Fig. 4b). Except for P. trichocarpa, GIP1 orthologs contained 1–5 Cys residues and the cysteine at position 45 in Arabidopsis GIP1 protein is conserved in the different orthologs suggesting a similar regulatory function of GIP1 proteins in other plants; however, this needs further investigation.
GIP1 was isolated as ZmGBF1 interacting protein in a yeast two-hybrid screen using A. thaliana cDNA library (Sehnke et al. 2005). It was proposed as a nuclear chaperone or crowbar that potentially regulates the multimeric state of GBFs (Sehnke et al. 2005). Within the cell, molecular chaperones can form oligomeric complexes of up to 1 MDa (Arrigo et al. 1988). Under non-reducing conditions, GIP1 revealed high-molecular weight oligomers in the MDa range. Several oligomers of GIP1 are likely formed through disulfide bonds (Fig. 5a, b). DTT results in significant reduction of GIP1 oligomers. However, oligomers formed by other means, e.g., hydrophobic interactions might exist (Fig. 5b bottom). In vitro, molecular chaperones act by preventing unfolded proteins from irreversible aggregation and, in cooperation with other factors like HSPs and ATP, facilitating productive refolding of unfolded proteins (Ehrnsperger et al. 1997; Lee et al. 1997). However, under non-reducing conditions, GIP1 possesses chaperone activity in an ATP-independent manner (Sehnke et al. 2005). The chaperone activity of GIP1 seems to depend on its oligomeric conformation (Fig. 7). This chaperone activity is lost with the reduced form of GIP1 (Fig. 7).
Intracellular redox potential has been proposed as a mechanism for the regulation of the activity of many proteins and dithiol/disulfide exchange is considered a key component of the redox-dependent transcriptional regulatory mechanisms (Dietz 2003; Liu et al. 2005). DNA-binding activity of plant transcription factors have been shown to be directly (Torn et al. 2002; Heine et al. 2004; Shaikhali et al. 2008; Stroher et al. 2009; Klein et al. 2012) or indirectly (Despres et al. 2003) regulated by redox. A novel redox-regulated mechanism has been proposed for members of the G-group bZIP transcription factors AtbZIP16, AtbZIP68, and AtGBF1 (Shaikhali et al. 2012). Although DNA-binding activity of the three TFs is greatly enhanced by reducing agents such as DTT (Shaikhali et al. 2012), their physiological reducing agents still to be identified. In mammals, yeast and bacteria thioredoxins and glutaredoxins act as possible physiological reducing agents for TFs (Matthews et al. 1992; Izawa et al. 1999; Zheng et al. 1998). This study shows that another level of redox regulation of these three TFs takes place through GIP1. It is interesting that reduced GIP1 enhanced the DNA-binding activity of the three TFs. Two possibilities for the function of GIP1 on bZIPs 1) through directly acting as reducing factor like Txs and Grxs or 2) GIP1 might act as an intermediate between the reducing factor and the TF like in the case of Ref1 (Wei et al. 2000).
Multiple functions of GIP1 seems to be associated with its oligomeric states that reversibly switch from LMW to HMW complexes and vice versa in response to the redox state of the cell (Fig. 9a). Oligomerization dependent functional changes have also been observed in human G3PD. Depending on its oligomeric status, G3PD changes its function from a dehydrogenase to a nuclear uracil-DNA glycosylase (Meyer-Siegler et al. 1991). Significant reduction in the DNA-bZIP complexes that occurs upon addition of non-reduced GIP1 suggests that specific oligomeric forms of GIP1 affect the DNA-binding capacity of these transcription factors through either interaction with the bZIP protein or the association with the DNA-protein complex (Fig. 6). As described in Fig. 9a, the hypothetical model suggests a mechanistic understanding of two phenomena. First, how the GIP1-bZIP interaction might be regulated in response to the redox potential of the cell; second, its effect on the regulation of DNA binding activity by these transcription factors in situations, i.e., light signaling, such as during photomorphogenesis.

Models of GIP function. a Hypothetical model depicting chaperon function of GIP1 and redox control of GIP1 on DNA-binding activity of members of the G-group bZIP transcription factors. 1 bZIPs binds DNA as a dimer. 2 In the cell when the redox potential is highly reduced GIP1 becomes reduced (monomerizes) and adopts a conformation that allows interaction/association with bZIP and/or bZIP-DNA complex which enhances DNA binding activity to the cognate cis-element. 3 under non-reducing conditions/slight oxidation by air, several oligomeric forms of GIP1 exist. Either association of particular oligomer to the bZIP-DNA complex or competition of different oligomers for such association results in disturbing the DNA-protein complex thus reducing the DNA binding activity. 4 Oxidation or highly oxidized redox potential leads GIP1 to form high molecular weight oligomer which prevents interaction/association with bZIP and/or bZIP-DNA complex. In the absence of GIP1 interaction, the binding of bZIPs to the DNA is not changed. In response to redox, there is a functional switching from redox to chaperon activity. b A working model shows red and blue light-specific regulatory roles of GIP1 and members of the G-group bZIP transcription factors in Arabidopsis early seedling development
Indeed GBF1 functions as negative regulator in blue light-mediated inhibition of hypocotyl elongation and as positive regulator of cotyledon expansion (Mallappa et al. 2006); however, bZIP16 functions as negative regulator in red light-mediated inhibition of hypocotyl elongation and as positive regulator of seed germination during photomorphogenesis (Hsieh et al. 2012). Besides GBF1, bZIP68 functions also as negative regulator in blue light-mediated inhibition of hypocotyl elongation (Shaikhali, unpublished data). Moreover, transgenic Arabidopsis lines overexpressing the wild-type version of bZIP16 and T-DNA insertion mutants for bZIP68 and GBF1 demonstrated impaired regulation of LHCB2.4 expression in response to light suggesting that bZIP68 and GBF1 function as transcriptional activators while bZIP16 functions as transcriptional repressor during photomorphogenesis (Shaikhali et al. 2012). Co-expression analysis of GIP1, bZIP16, bZIP68, and GBF1 genes in response to red and blue light and seed germination after 12 and 24 h revealed correlations for GIP1 and bZIP16 and for GIP1, bZIP68, and GBF1 (Fig. S2B) indicating that GIP1 functions as co-regulator of these transcription factors for their specific responses during photomorphogenesis. When the early light response was investigated in the 35S:GIP1 overexpression lines, impaired induction of LHCB2.4 expression compared with wild type was observed. In the 35S:GIP1 overexpression lines, the levels of LHCB2.4 transcript were significantly reduced compared with WT, suggesting that GIP1 acts as a co-repressor of LHCB2.4. Expression of LHCB2.4 is strongly repressed in response to redox changes in the chloroplast (Kleine et al. 2007), and bZIP16 possibly plays a role in mediating this repression (Shaikhali et al. 2012). In support of the gene expression data and of the proposed role for GIP1 as a co-repressor of photosynthetic gene expression, the 35S:GIP1 over-expression lines demonstrated elongated hypocotyls compared with WT in response to RL (Figs. 8 and 9), an effect observed earlier for 35S:bZIP16 overexpression line (Hsieh et al. 2012). GIP1 functions not only a transcriptional co-repressor of LHCB2.4 gene expression in response to RL but also as a transcriptional co-activator in response to BL (Figs. 8 and 9). Again, in support of the gene expression data and of the proposed role for GIP1 as a co-activator of photosynthetic gene expression, the 35S:GIP1 overexpression lines demonstrated elongated hypocotyls compared with WT in response to BL (Figs. 8 and 9). T-DNA insertion lines for bZIP68 and GBF1 also demonstrated reduced LHCB2.4 expression levels in response to BL and suggested that bZIP68 and GBF1 function as activators for LHCB2.4 expression (Shaikhali et al. 2012; Mallappa et al. 2006). In addition, 35S:GBF1 overexpression line has also been shown to exhibit elongated hypocotyls in response to white and blue light (Mallappa et al. 2006). It is clear that GIP1 forms heterodimers with bZIP16, bZIP68, and GBF1 (Fig. 2). Heterodimer formation increases the diversity of functional G-box binding combinations and depending on the recruited partner, a different output is generated. DNA binding affinity and specificity, transactivation potential, and overall cell physiological function have been suggested to be altered by heterodimerization (Naar et al. 2001). Thus, our results suggest that combinatorial interactions among GIP1, bZIP16, bZIP68, and GBF1 play a role in generating different light signaling outputs enabling a redox-controlled response to changes in the environment.
Abbreviations
- BL:
-
Blue light
- BN:
-
Blue native
- bZIP:
-
Basic region leucine zipper
- CLSM:
-
Confocal laser scanning microscopy
- DTT:
-
Dithiothreitol
- EMSA:
-
Electrophoretic mobility shift assay
- GIP1:
-
GBF1 interacting protein 1
- IOD:
-
Iodoacetamide
- LBD18:
-
Lateral organ boundaries domain 18
- NEM:
-
N-ethylmaleimide
- NLS:
-
Nuclear localization signal
- NPE:
-
Nuclear protein extracts
- ONPG:
-
O-nitrophenyl β-d-galactopyranoside
- RL:
-
Red light
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TPE:
-
Total protein extraction
- 2D:
-
Two-dimensional
References
Ang LH, Deng XW (1994) Regulatory hierarchy of photomorphogenic loci: allele-specific and light-dependent interaction between HY5 and COP1 loci. Plant Cell 6:613–628
Arrigo AP, Suhan JP, Welch WJ (1988) Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol 8:5059–5071
Chen M, Chory J, Fankhauser C (2004) Light signal transduction in higher plants. Annu Rev Genet 38:87–117
Chi Y, Yang Y, Zhou Y, Zhou J, Fan B, Yu JQ, Chen Z (2013) Protein-protein interactions in the regulation of WRKY transcription factors. Mol Plant 6(2):287–300
Deppmann CD, Acharya A, Rishi V, Wobbes B, Smeekens S, Taparowsky EJ, Vinson C (2004) Dimerization specificity of all 67 B-ZIP motifs in Arabidopsis thaliana: a comparison to Homo sapiens B-ZIP motifs. Nucleic Acids Res 32:3435–3445
Despres C, Chubak C, Rochon A, Clark R, Bethune T, Desveaux D, Fobert PR (2003) The Arabidopsis NPR1 disease resistance protein is a novel cofactor that confers redox regulation of DNA binding activity to the basic domain/leucine zipper transcription factor TGA1. Plant Cell 15:2181–2191
Dietz KJ (2003) Redox control, redox signaling, and redox homeostasis in plant cells. Int Rev Cytol 228:141–193
Ehrnsperger M, Graber S, Gaestel M, Buchner J (1997) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 16:221–229
Eubel H, Braun HP, Millar AH (2005) Blue-native PAGE in plants: a tool in analysis of protein-protein interactions. Plant Methods 1:11
Foster R, Izawa T, Chua NH (1994) Plant bZIP proteins gather at ACGT elements. FASEB J 8:192–200
Heine GF, Hernandez JM, Grotewold E (2004) Two cysteines in plant R2R3 MYB domains participate in REDOX-dependent DNA binding. J Biol Chem 279:37878–37885
Holm M, Ma LG, Qu LJ, Deng XW (2002) Two interacting bZIP proteins are direct targets of COP1-mediated control of light-dependent gene expression in Arabidopsis. Gene Dev 16:1247–1259
Hsieh WP, Hsieh HL, Wu SH (2012) Arabidopsis bZIP16 transcription factor integrates light and hormones signaling pathways to regulate early seedling development. Plant Cell 24:3997–4011
Izawa S, Maeda K, Sugiyama K, Mano J, Inoue Y, Kimura A (1999) Thioredoxin deficiency causes the constitutive activation of Yap1, an AP-1-like transcription factor in Saccharomyces cerevisiae. J Biol Chem 274:28459–28465
Jakoby M, Weisshaar B, Droge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj T, Parcy F (2002) bZIP transcription factors in Arabidopsis. Trends Plant Sci 7:106–111
Jiao Y, Lao OS, Deng XW (2007) Light regulated transcriptional networks in higher plants. Nat Rev Genet 8:217–230
Klein P, Seidel T, Stöcker B, Dietz KJ (2012) The membrane-tethered transcription factor ANAC089 serves as redox-dependent suppressor of stromal ascorbate peroxidase gene expression. Front Plant Sci 3:247
Kleine T, Kindgren P, Benedict C, Hendrickson L, Strand Å (2007) Genome-wide gene expression analysis reveals a critical role for CRYPTOCHROME1 in the response of Arabidopsis to high irradiance. Plant Physiol 144:1391–1406
Lee GJ, Roseman AM, Saibil HR, Vierling E (1997) A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding component state. EMBO J 16:659–671
Lee HW, Park JH, Park MY, Kim J (2014) GIP1 may act as a coactivator that enhances transcriptional activity of LBD18 in Arabidopsis. J Plant Physiol 171:14–18
Liu L, White MJ, MacRae TH (1999) Transcription factors and their genes in higher plants: functional domains, evolution and regulation. Eur J Biochem 262:247–257
Liu H, Colavitti R, Rovira II, Finkel T (2005) Redox-dependent transcriptional regulation. Circ Res 97:967–974
Ma L, Li J, Qu L, Hager J, Chen Z, Zhao H, Deng XW (2001) Light control of Arabidopsis development entails coordinated regulation of genome expression and cellular pathways. Plant Cell 13:2589–2607
Mallappa C, Yadav V, Negi P, Chattopadhyay S (2006) A basic leucine zipper transcription factor, G-box-binding factor 1, regulates blue light-mediated photomorphogenic growth in Arabidopsis. J Biol Chem 281:22190–22199
Mallappa C, Singh A, Ram H, Chattopadhyay S (2008) GBF1, a transcription factor of blue light signaling in Arabidopsis is degraded in the dark by a proteasome mediated pathway independent of COP1 and SPA1. J Biol Chem 283:35772–35782
Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT (1992) Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 20:3821–3830
Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, Dierel JK, Sirover MA (1991) A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci U S A 88:8460–8464
Naar AM, Lemon BD, Tjian R (2001) Transcriptional coactivator complexes. Annu Rev Biochem 70:475–501
Nagatani A, Reed JW, Chory J (1993) Isolation and initial characterization of Arabidopsis mutants that Are deficient in phytochrome A. Plant Physiol 102:269–277
Neff MM, Fanhauser C, Chory J (2000) Light: an indicator of time and place. Gen Dev 14:257–271
Pepper AE, Chory J (1997) Extragenic suppressors of the Arabidopsis det1 mutant identify elements of flowering time and light response regulatory pathways. Genetics 145:1125–1137
Ron D, Habener JF (1992) CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant negative inhibitor of gene transcription. Gene Dev 6:439–453
Schindler U, Menkens AE, Beckmann H, Ecker JR, Cashmore AR (1992) Heterodimerization between light-regulated and ubiquitously expressed Arabidopsis GBF bZIP proteins. EMBO J 11:1261–1273
Sehnke PC, Laughner BJ, Linebarger CL, Gurley WB, Ferl RJ (2005) Identification characterization of GIP1, an Arabidopsis thaliana protein that enhances the DNA binding affinity and reduces the oligomeric state of G-box binding factors. Cell Res 15:567–575
Shaikhali J, Heiber I, Seidel T, Stroher E, Hiltscher H, Birkmann S, Dietz KJ, Baier M (2008) The redox-sensitive transcription factor Rap2.4a controls nuclear expression of 2-Cys peroxiredoxin A and other chloroplast antioxidant enzymes. BMC Plant Biol 8:48
Shaikhali J, Noren L, Barajas-Lopéz JD, Srivastava V, König J, Sauer UH, Wingsle G, Dietz KJ, Strand A (2012) Redox-mediated mechanisms regulate DNA-binding activity of the G-group of bZIP transcription factors in Arabidopsis. J Biol Chem 287:27510–27525
Shen H, Cao K, Wang X (2008) AtbZIP16 and AtbZIP68, two new members of GBFs, can interact with other G group bZIPs in Arabidopsis thaliana. BMB Rep 41:132–138
Siberil Y, Doireau P, Gantet P (2001) Plant G-box binding factors: modular structure and activation mechanisms. Eur J Biochem 268:5655–5666
Stroher E, Wang XJ, Roloff N, Klein P, Husemann A, Dietz KJ (2009) Redox-dependent regulation of the stress-induced zinc-finger protein SAP12 in Arabidopsis thaliana. Mol Plant 2:357–367
Su PH, Li HM (2008) Arabidopsis stromal 70-kD heat shock proteins are essential for plant development and important for thermotolerance of germinating seeds. Plant Physiol 146:1231–1241
Tada Y, Spoel SH, Pajerowska-Mukhtar K, Mou Z, Song J, Wang C, Zuo J, Dong X (2009) Plant immunity requires conformational changes of NPR1 via S-nitrosylation and thioredoxins. Science 321:952–956
Tamai H, Iwabuchi M, Meshi T (2002) Arabidopsis GARP transcriptional activators interact with the Pro-rich activation domain shared by G-box-binding bZIP factors. Plant Cell Physiol 43:99–107
Tandon S, Horowitz PM (1989) Reversible folding of rhodanese. Presence of intermediate(s) at equilibrium. J Biol Chem 264:9859–9866
Torn AE, Bertoncini CW, Chan RL, Gonzales DH (2002) Redox regulation of plant homeodomain transcription factors. J Biol Chem 277:34800–34807
Vincentz M, Bandeira-Kobarg C, Gauer L, Schlögl P, Leite A (2003) Evolutionary pattern of angiosperm bZIP factors homologous to the maize Opaque2 regulatory protein. J Mol Evol 56:105–116
Virbasius CM, Wagner S, Green MR (1999) A human nuclear-localized chaperone that regulates dimerization, DNA binding and transcriptional activity of bZIP proteins. Mol Cell 4:219–228
Von Arnim A, Deng XW (1996) Light control of seedling development. Annu Rev Plant Physiol Plant Mol Biol 47:215–243
Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D (2000) Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res 60:6688–6695
Whitelam GC, Johnson E, Peng J, Carol P, Anderson ML, Cowl JS, Harberd NP (1993) Phytochrome A null mutants of Arabidopsis display a wild-type phenotype in white light. Plant Cell 5:757–768
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Zheng M, Aslund F, Storz G (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279:1718–1721
Acknowledgments
I would like to thank Prof. Gunnar Wingsle and Dr. Juande Dios Barajas-Lopez for their critical reading of the manuscript. I thank Dr. Robert J. Ferl for the gift of anti-GIP1 antibody. Dr. Jungmook Kim is gratefully acknowledged for providing seeds of 35S:GIP1 transgenic lines. This work was supported by a grant from the KEMPE foundation.
Conflict of interest
The author declares that he has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Peter Nick
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Homology of ZmGBF1, AtGBF3 and AtGBF1 proteins. A, Amino acid sequence alignment of ZmGBF1, AtGBF3 and AtGBF1 proteins. The asterisks (*) indicates perfectly conserved amino acid residues, (:) indicates general similarities, similarity among 2 to 4 is represented by (.) and dashes (−) indicate gaps introduced to maximize alignment. B, The phylogenetic tree was produced using Phylogeny.fr (http://www.phylogeny.fr/). (PDF 149 kb)
Figure S2
Gene expression of GIP1, bZIP16, bZIP68 and GBF1 genes during development and photomorphogenesis. A, Transcript abundance in the different stages of Arabidopsis development using the developmental tool of Genevestigator (www.genevestigator.com). B, Heat map representing the co-expression patterns of GIP1, bZIP16, bZIP68 and GBF1 genes (in row) during photomorphogenesis such as seed germination after 12 and 24 h and in response to red and blue light (in column). (PDF 84 kb)
Rights and permissions
About this article

Cite this article
Shaikhali, J. GIP1 protein is a novel cofactor that regulates DNA-binding affinity of redox-regulated members of bZIP transcription factors involved in the early stages of Arabidopsis development. Protoplasma 252, 867–883 (2015). https://doi.org/10.1007/s00709-014-0726-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-014-0726-9