
Abstract
Herein, we have synthesized four indolo[2,3-b]quinoxalin-2-yl)(phenyl)methanone derivatives by cyclocondensation. The photophysical studies of dyes in various solvents and neat solid film exhibit typical electronic spectra with inbuilt intramolecular charge transfer (ICT) (λmax: 397–490 nm) confirming donor–acceptor architecture. Herein, dyes fluoresce in the blue-orange region (λEmax: 435–614) on excitation at their ICT maxima in toluene, ethyl acetate, chloroform, DMSO, and neat solid film. Two dyes, which exhibit good emission intensity in all mediums, were studied for aggregation-induced emission (AIE) effect. Electrochemical studies indicate the dyes possess relatively low lying LUMO (− 3.65 to − 3.98 eV) comparable to reported n-type/electron-transporting materials. The HOMO and LUMO energy levels were evaluated by DFT and TD-DFT calculations. TGA analysis shows the dyes exhibit good thermal stability. The characteristic optoelectronic properties and thermal stability signify these dyes are potential candidates for their application in optoelectronics.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Organic materials are emerging as third-generation materials due to their widespread applications in organic light-emitting diodes (OLEDs) [1, 2], organic field-effect transistors (OFETs) [3], dye-sensitized solar cells (DSSCs) [4,5,6], non-linear optics (NLO’s) [7], chemo/biosensors [8, 9], etc. Researchers are making effort to further improve the efficiency and lifetime of organic materials, to exploit their hidden potential. As organic materials offer low cost and easy device fabrications. In particular, the donor–acceptor (D‒A) based design approach which enables simpler tuning of optoelectronic properties with molecular engineering, was found to be promising to obtain organic material with diverse properties such as solvatofluorochromism, two-photon excited fluorescence, triplet–triplet annihilation (TTA), thermally activated delayed fluorescence (TADF), and charge transport (hole/electron). However, the fluorescence of many organic fluorophores which emit brightly in the solution state get diminishes in the solid/aggregate state due to aggregation-caused quenching (ACQ). Indeed, it is desirable to have molecules emit in a solid/aggregate state for their real-world application. In 2001, Tang et al. first introduced aggregation-induced emission (AIE) which is contradictory to detrimental ACQ [10]. The twist in molecular structure, attachment of bulky donor groups, or incorporation of AIEgens (e.g., tetraphenylethene (TPE), hexylphenylsilole (HPS)) to ACQ fluorophores (anthracene, pyrene, PBI, BODIPY, TPA, carbazole, etc.) can be an effective approach to obtain AIE active molecules [11, 12].
Additionally, the charge transport in an organic molecule is very important to realize for their application in devices. Organic materials should have HOMO/LUMO energy levels comparable to the work function of commonly used electrodes (anode: ITO and cathode: Ca/Al/Mg) to facilitate charge transport [13]. Organic semiconducting molecules which have HOMO (LUMO) levels at around 5.0 (4.0) eV are capable of hole (electron) transport and known as hole (electron)-transporting or p (n)-type materials [14, 15]. Compared to p-type, the development of n-type materials has lagged due to their difficult synthesis and instability under ambient conditions (in presence of H2O/O2). This raises the need for air-stable n-type materials as it is an integral part of the complementary circuit and received unprecedented attention from researchers. Though there is no general guideline to obtain air-stable n-type materials, according to the literature LUMO level can be lowered through the introduction of electron-withdrawing groups ‒F, ‒CN, ‒Cl, ‒NO2, ‒COOH, ‒CF3, and perfluorinated alkyl chain in p-type materials. An alternative approach is the incorporation of electronegative heteroatoms like N or S to acceptor core of D-A system can promote injection and transport of electrons in resulting molecules [16]. Previously, we designed and reported different D‒A based molecules comprised of donor moieties like aromatic amines (triarylamine/diarylamine/aryl), heterocyclic amine (carbazole/phenothiazine), and acceptor moieties quinoxaline [17,18,19,20,21], indoloquinoxaline [22, 23], phenazine [24,25,26], pyrido-pyrazine [27, 28], pyrazino-phenazines [29], acridone [30], and anthraquinone [31], to obtain a broad range of emission, solvatofluorochromism, AIE, TADF, hole/electron/bipolar charge transport for their application in organic electronics.
Indoloquinoxaline (IQ) is also one of the important classes of N-containing heterocyclic compounds. Compared to organic electronics, indole and its IQ derivatives have been extensively studied in bio/medical chemistry for two reasons (i) as it shows the wide spectrum of biological activity such as anti-viral [32, 33], anti-cancer [34, 35], anti-microbial [36], anti-bacterial agents [37], anti-HIV and so on [38]. (ii) Indole is a chromophore of important amino acid, tryptophan [39]. Along with the importance of luminescence of indole in tryptophan, literature also witnesses complexity in emission spectra of indole derivatives in various solvents due to the presence of two degenerate excited states La and Lb differing in dipole moment demonstrating sensitivity towards solvent polarity [39,40,41].
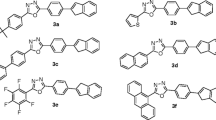
Encouraged by previous work, and looking at the scope of IQ materials in organic electronics, we present the design and synthesis of four indoloquinoxaline (IQ) derivatives, based on inbuilt D‒A architecture in which electron-rich indole act as a donor and electron-deficient quinoxaline act as an acceptor. Further, the electron deficiency of quinoxaline has been increased through the introduction of the benzoyl segment. The presence of the benzoyl group is not only useful to lower the LUMO level but also responsible to bring some twist in the molecule so that detrimental π–π stacking in solid-state get minimized. The influence of electron-donating (–CH3) and withdrawing substituents (–Br, –NO2) and increased electron deficiency of acceptor core through benzoyl unit, on opto-electrochemical properties have been studied. The detailed photophysical, AIE, electrochemical, thermal, and theoretical properties of synthesized dyes are investigated. The molecular structure of IQ derivatives is presented in Fig. 1.

Synthesized indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone derivatives 1–4
Result and discussion
Synthesis and characterization
Four indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone derivatives based on D‒A architecture have been synthesized through a simple cyclo-condensation reaction, The synthesis of target compounds is shown in Scheme 1. IQ derivatives 1–4 were synthesized in two steps. Firstly, isatin derivatives were alkylated at their N-position to increase the solubility of the target molecules, and later these N-methylated isatin derivatives were reacted with 1,2-diaminobenzophenone in acetic acid for 3–4 h to produce desired cyclo-condensed products 1–4. The four target compounds were obtained in good yield (40–85%) as yellow–brown solids. The identity and purity of all new compounds were confirmed by several characterization methods such as FT-IR, 1H NMR, 13C NMR, MALDI-TOF, etc. The inbuilt D‒A architecture and HOMO–LUMO orbital energies were further rationalized using DFT calculations.

Photophysical properties
First, photophysical properties of 1–4 were studied in increasing polarity of solvents viz., toluene, ethyl acetate, chloroform, and DMSO to understand the effects of solvent polarity on spectral profiles of dyes and have correlated it with their structures. The pertinent data are listed in Table 1 and absorption spectra of 1–4 recorded in solution are presented in Fig. 2.

Absorption spectra of compounds 1–4 in toluene (a), ethyl acetate (b), chloroform (c), and dimethyl sulfoxide (d)
The absorption spectra of 1–4 in different solvents are dominated by multiple overlapping bands assigned to different chromophoric segments in the molecule where, the intense bands in the region 261–404 nm corresponds to high energy n‒π* and π‒π* transitions in the molecules. The characteristic dual absorptions band between 324 and 369 nm suggests the occurrence of π‒π* transitions from S0 to S2 and S1 state, respectively which could arise from the indole segment of the molecule [42]. Additionally, all derivatives exhibit a lower energy transition around 397–490 nm that can be assigned to inbuilt intramolecular charge transfer (ICT) from electron-donating indole unit to electron-deficient quinoxaline core. These ICT transitions were bathochromically shifted for derivatives 2–4 substituted with electron-donating/withdrawing group compared to 1.
The ICT band in 3 is most red-shifted with 18–81 nm and covers a broad range (up to 600 nm) suggests an increased electron-donating strength of indole due to ‒Br substituent (+ M mesomeric effect). Besides this, high-intensity ICT transitions in 4 indicate better charge separation and charge transfer in the D–A system due to the presence of electron-withdrawing ‒NO2 (–M effect) on indole moiety. Dyes display good molar absorptivity and almost similar absorption spectra with varying intensities in different solvents. The longer wavelength ICT band seems slightly sensitive toward the nature of solvents. The optical band gap of compounds estimated from the offset wavelength of the low energy absorption band lies within the range of 2.08–2.70 eV in solution (Table 1).
Thereafter, in the neat solid film the absorption spectra of 1–4 (Fig. 4a) displayed a broadening of bands with a significant redshift of 8–104 nm in absorption maxima attributed to the molecular aggregation.
The other photophysical data such as optical band gap, emission data, Stoke’s shift, and quantum yield were also calculated and summarized in Table 1. Dyes 1–4 emit blue-orange fluorescence with emission maxima λEmax: 458–614 nm on excitation at their respective ICT maxima in toluene, ethyl acetate, chloroform, DMSO, and neat solid film (Figs. 3 and 4A). The quantum yield of 1–4 was calculated using fluorescein (Ф = 0.79 in 0.1 N NaOH) as a reference and are in the range 0.01–0.23 in toluene.

Emission spectra of compounds 1–4 in toluene (a), ethyl acetate (b), chloroform (c), and dimethyl sulfoxide (d)

Absorption spectra (a) and emission spectra (A) of 1–4 in a neat solid film
Dyes 1 and 2 display detectable emissions in toluene, ethyl acetate, and chloroform whereas weak emissions obtained for dyes 3 and 4 in toluene get considerably quenched in more polar solvents except in chloroform. This reduced intensity or quenching of emission might be due to the presence of ‒Br in 3 and strong electron-withdrawing ‒NO2 in 4.
On contrary, the ability of H-bonding bonding by ‒Br and ‒NO2 group present in 3 and 4 with chloroform is responsible for improved long-wavelength emission at λEmax: 582 nm and λEmax: 544 nm, respectively. Further, all dyes in more polar solvent DMSO displays quenching in emission with featureless emission spectra indicating the occurrence of non-radiative relaxation of the excited state through dipole–dipole interaction. Intriguingly, the bathochromic shift in the emission of dyes with the decrease in emission intensity from toluene (non-polar) (λEmax: 435–508 nm) to chloroform (λEmax: 443–582 nm) and quenching of emission in polar DMSO demonstrate the sensitivity of dyes towards polarity of the solvent. (See Supporting information (SI), Fig. S1, for the enlarged emission spectra in the above-mentioned solvents.)
Nonetheless, emission maxima in the different solvent, ICT absorption band, and Stoke’s shift of dyes in solution suggests the existence of specific solute–solvent interaction [43]. In toluene, derivative 3 emits at a shorter wavelength (λEmax: 435, 460 nm) compared to ICT absorbance maxima (λICTmax: 489 nm). Though the emission intensity is low, the observed shorter wavelength emission and longer wavelength-broad ICT absorbance band with anti-Stoke’s shift in 3 is probably the consequence of the occurrence of hot-band absorption or some up-conversion process in a molecule. In hot-band absorption, the molecule absorbs from a higher vibrational level (hot band) of the ground state thus resulting in a longer wavelength (lower energy) absorbance band and emission obtained is the usual fluorescence process that gives anti-Stoke’s shift. However, in the up-conversion process, the excited state ion absorbs energy from another excited state ion (or sensitizer) and reaches to higher excited state that leads to high energy emission with anti-Stoke’s shift. Luminescent materials possessing anti-Stoke’s shifts are useful in bio-applications [44].
Furthermore, dyes 1 and 2 display more red-shifted emission and greater Stoke’s shift in ethyl acetate than chloroform verifying specific solute–solvent interaction in ethyl acetate. This specifies more stabilization of the excited state of dye with orientation polarizability of ethyl acetate. Excluding the exception of anti-Stoke’s shift observed for dye 3 in toluene, all dyes exhibit considerable Stoke’s shift in the range 1236–9364 cm−1 in solution and solid film which ensures reduction of reabsorption of the emitted photons.
Almost all dyes exhibit more or less emission in the neat solid film with a bathochromic shift of 12–79 nm compared to the solution. It could be attributed to intermolecular aggregation in the solid-state. Dyes 1 and 2 display better emission in solid-state reveals the capability of aggregation induce emission tested for AIE characteristic.
AIE phenomenon
In order to estimate the presence of AIE properties in dyes 1 and 2 at the preliminary stage, we have prepared a series of THF/water mixtures (0–100%) of 1 and 2 according to the reported procedure [45] to form nanoaggregates of organic dyes and observed them under UV light. The changes in fluorescence properties of dye with varying concentrations of water from 0 to 100% have been studied thoroughly and presented in Fig. 5 (for AIE of dye 2, see SI Fig. S2). This shows at higher water fractions quenching of luminescence is greatly recovered due to the presence of AIE activity.

Fluorescence image of 1 in THF—water mixture with subsequently increasing water fractions (10–5 M) by 10% up to 80% and by 5% from 80 to 100% under UV–Vis light (a); Fluorescence spectra of 1 in THF— water mixture (10–5 M) with increasing water fractions (ƒw) (b); plots of PL intensity and peak wavelength versus ƒw (c); and DLS plot of 1 obtained for nano-aggregates formed in fw = 95% of THF—water mixture (d)
Both the dyes exhibit yellow emission in pure THF solution that could be attributed to inbuilt ICT from donor indole unit to acceptor quinoxaline core. Further, the addition of water dramatically decreases the emission intensity of dye solution with a slight redshift from fw 10–80% and up to 70% in 1 and 2, respectively indicate the occurrence of the positive solvatochromism that could be a result of increased solvent polarity due to the addition of water. Thereafter, enhancement of water content onwards fw 80% in 1 and 70% in 2 greatly recovers the emission of dyes, implying the formation of nanoaggregates at higher water fraction. The formation of nanoaggregates restricts detrimental relaxation processes such as intramolecular rotations (RIR) and vibrations (RIV), opens up the radiative channel, and allows luminophore emits strongly [46]. Both the dyes solution depicts the highest luminescence at fw 95% with the relative intensity of ~ 1016 and ~ 136 in 1 and 2, a spectacle the presence of aggregation-induced enhanced emission (AIEE) and AIE, respectively. This more and less emission intensity of nanosuspensions corresponds to different physical constraints/modes of aggregation, effective electronic conjugation, and yield of nanoparticles [12]. The difficulty to control the size, shape, and yield of nanoparticle formation at higher water fractions usually results into an irregular pattern of AIE intensity [12, 47]. The level off tail or light scattering at higher water fraction mixtures in absorption spectra is due to the formation of nanoaggregates in suspension. This phenomenon is also known as “Mie scattering” [48] (For Absorption Spectra, See SI Fig. S3). The formation and stability of nanosuspensions were further examined and confirmed by DLS, which helped to relate emission intensity with the size and stability of nanosuspensions. The highest emission intensity obtained at fw 95% for both dyes 1 and 2 is the result of the smallest hydrodynamic size and low polydispersity index (PDI < 0.5) of nanoaggregates. It shows emission intensity is inversely proportional to the hydrodynamic size of nanosuspension and directly proportional to the stability of nanoaggregates.
Electrochemical properties
The electrochemical behavior of dyes and their energy level were identified by cyclic voltammetry. The experiment was performed in anhydrous dichloromethane with tetrabutylammonium hexafluorophosphate (Bu4NPF6) as electrolyte and ferrocene as an internal standard to calibrate redox potential. The cyclic voltammogram of dye 3 and graph of optical vs electrochemical bandgap of 1–4 are presented in Fig. 6 and the pertinent data are preserved in Table 2 (for CV of other compounds, see SI Fig. S4).

Cyclic voltammogram (full scan) of dye 3 (a) in anhydrous dichloromethane and graph of optical vs. electrochemical bandgap (b)
On an anodic sweep, a noticeable irreversible oxidation peak observed in 3 corresponds to the oxidation of indole segment in the molecule, whereas no proper oxidation peak has been detected in 1, 2, and 4 due to the low intensity of the oxidation wave. Further, the cathodic sweep of 1–4 predominately exhibits two quasireversible waves corresponding to the reduction of quinoxaline unit and carbonyl group in the benzoyl arm of the synthesized molecules. In addition to this, one extra quasireversible reduction peak obtained for derivative 4 can be assigned to the reduction of electron-withdrawing ‒NO2 group present on indole moiety.
The HOMO and LUMO energy levels of organic materials are the crucial parameters for their application in optoelectronic devices. Thus, to estimate HOMO and LUMO energy levels and to understand the type of charge transport in the molecules, the LUMO energy level of 1–4 was calculated by using the first reduction potential whereas the HOMO level of all dyes except 3 was calculated using the difference of optical band gap and LUMO energy values.
The first oxidation potential obtained in the cyclic voltammogram of 3 has been used to calculate its HOMO level. The calculated HOMO and LUMO energy values of 1–4 fall within the range − 5.64 to − 6.35 eV and − 3.65 to − 3.98 eV, respectively. Among these dyes, ‒NO2 substituted indoloquinoxaline derivative 4 possesses the lowest LUMO (− 3.98 eV) may be due to the increased electron-accepting strength of the electron-withdrawing effect of ‒NO2. Thereafter, ‒Br substituted derivative 3 shows low LUMO (− 3.72 eV) with optimized HOMO (− 5.64 eV) compared to unsubstituted derivative 1 indicate the influence of -I and mesomeric effect (+ M effect) of ‒Br group on the energy level of the molecule. The LUMO energy levels of 1–4 is comparable with reported n-type materials perfluoroalkyl substituted C60-fused N-methylpyrrolidine-para-dodecyl phenyl derivative (C60PC12F25) (LUMO = − 3.63 eV) [49], N,N′-bis(1H,1H,2H,2H-perfluorodecyl)-1,4,5,8-naphthalenetetracarboxylic diimide [50], and pyromellitic diimide derivatives [51] indicate their capability of electron transport. The electrochemical bandgap of 1–4 is in the range of 1.92–2.70 eV and comparable with the optical band gap (Fig. 6b).
Theoretical properties
Quantum chemistry calculations are performed using Gaussian03 within the framework of density functional theory (DFT). The final geometries for each molecule were obtained by optimizing the initial structure in the gas state within the approximation of B3LYP hybrid exchange–correlation functional and 6–311 + + G** basis set [52]. The optimized structure, electronic distribution in the frontier molecular orbitals, and absorption spectra of 1 in the gas phase and different solvents (toluene, chloroform, and DMSO) are presented in Fig. 7 (for other compounds, see SI Fig. S17-S25).

Optimized structure (a) and frontier molecular orbitals (b) of 1 in the gas phase; and simulated absorption spectra of 1 in gas, toluene, CHCl3, and DMSO (c)
Though HOMO and LUMO orbitals of derivatives are overlapped, it can be distinguished that the HOMO orbital presides mainly on electron-rich indole and the LUMO orbital strongly resides on accepting moieties like quinoxaline and carbonyl. The location of HOMO (LUMO) orbitals on the indole (quinoxaline) features the inbuilt donor–acceptor architecture in derivatives.
The first ionization potential, electron affinity, HOMO–LUMO levels, bandgap, and ground-state dipole moment were computed for the dyes 1–4 and given in Table 3. The HOMO (LUMO) energy of 1–4 is in the range of − 6.05 to − 6.70 eV (− 2.50 to − 2.99 eV), which are comparable with the experimentally observed energy levels. The dipole moments in the range 5.01–9.18 Debye validates the occurrence of charge transfer within the molecule. The large dipole moment (9.18 eV) in 4, due to the presence of the electron-withdrawing ‒NO2 group increases the charge separation hence improving the charge transfer.
The bandgap of molecules in the range 3.53–3.71 eV, exhibits a substitution effect. The small band gap in 2 (3.55 eV) and 3 (3.53 eV) could be attributed to electron-donating or + I effect of ‒CH3 and + M effect of ‒Br substituent, in comparison to 1. While strong electron-withdrawing/-M effect of ‒NO2 group is responsible for a larger bandgap in 4 (3.71 eV).
Finally, the optical properties were evaluated by TD-DFT in the gas phase, toluene, chloroform, and DMSO using the same parameters. The transitions above 350 nm correspond to inbuilt charge transfer in the dyes and were found to be affected by modulating peripheral electron-donating/withdrawing substituent. (See SI, Table S5‒S20) Similar to experimentally observed absorption spectra, the computationally obtained λmax are slightly sensitive towards solvent polarity (toluene < CHCl3 < DMSO (see SI, Figure S25). The Cartesian coordinates of optimized structures, Mulliken and Lowdin charges of 1–4 are given in SI Tables S1-S4.
Thermal properties
The thermal stability of 1–4 was studied via thermogravimetric analysis (TGA) and the melting point was determined by the open capillary method. The TGA thermogram of 1–4 has been obtained in a nitrogen atmosphere at normal pressure with a heating rate of 10 °C/min and is displayed in Fig. 8. The slight increase in mass on the heating cycle at beginning of the TGA thermogram can be attributed to the buoyancy effect [53].

TGA thermogram of 1–4 under nitrogen atmosphere at normal pressure. Heating rate 10 °C/min
TGA thermogram indicates that compounds 1–4 have good thermal stability. The decomposition temperature corresponds to 5% and 10% weight losses found in the range 261–314 °C and 286–335 °C, respectively (Table 4). The order of thermal stability in derivatives 1–4 is 4 > 3 > 2 > 1.
Conclusion
Indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone derivatives based on D‒A architecture are synthesized through a simple cyclocondensation reaction. The inbuilt D‒A architecture generates an ICT transition in the absorption spectrum of the molecule, which is noticeably influenced by electron-donating or withdrawing substituents present at the 9th position of indole moiety. Dyes emit blue-orange fluorescence (435–614 nm) on excitation at ICT maxima in toluene, ethyl acetate, chloroform, DMSO, and neat solid film. Emission maxima, ICT absorption band, and Stoke’s shift of dyes reveal the existence of specific solute–solvent interaction.
The fluorescence study of dyes 1 and 2 in different THF/water mixtures demonstrate the presence of AIEE in 1 and AIE in 2 with the formation of nano-aggregates, confirmed by DLS. Dyes exhibit low lying LUMO (− 3.65 to − 3.98 eV) which are comparable with reported n-type materials and display electron-transporting/injecting properties in 1–4. Thermal properties reveal that the derivatives have a high melting point and good thermal stability. DFT and TD-DFT studies verify the inbuilt donor–acceptor architecture of 1–4. The results indicate that synthesized compounds are potential candidates to be either used as n-type material or solid-state emitters in organic electronics.
Experimental
All the starting materials and reagents were purchased from commercial sources (Sigma Aldrich and Alfa Aesar) and were used without any further treatment and purification unless otherwise mentioned. The organic solvents were of HPLC and spectroscopic grade and were dried and freshly distilled using the standard procedures and handled in a moisture-free atmosphere. Column chromatography was carried out using SD-fine silica gel (60–120 mesh), eluting with n-hexane and chloroform. The progress of the reaction and the purity of the compound was checked by thin-layer chromatography (TLC) on silica-gel-coated glass plates, in which the spots were visualized with UV light (365 nm) and in an iodine chamber.
UV–Vis spectra were recorded in 10‒6 mol dm‒3 solution in a 1 cm path length quartz cuvette as well as the neat solid films on a SHIMADZU UV-2401PC instrument at room temperature. The neat solid films of compounds 1–4 were prepared by using a spin coater (Holmarc HO-TH-05) at 1000 rpm for 2 min ∼6 mg cm‒3 of the sample in chloroform. Quartz plates were used for neat solid film studies. The excitation and emission spectra were carried out on a Perkin Elmer LS 55 Fluorescence spectrophotometer. Cyclic voltammetry studies were carried out on a computer-controlled Palmsens3 potentiostat. Typically, a three-electrode cell equipped with a glassy carbon working electrode, an Ag/AgCl (non-aqueous) reference electrode, and platinum (Pt) wire as the counter electrode was employed. The measurements were carried at room temperature in anhydrous dichloromethane with tetrabutylammonium hexafluorophosphate solution (0.1 M) as the supporting electrolyte with a scan rate of 100 mV s‒1. The potential of the Ag/AgCl reference electrode was calibrated by using a ferrocene/ferrocenium redox couple, which has a known oxidation potential of + 5.1 eV. The thermogravimetric analysis (TGA) was performed using a Mettler Toledo instrument (TG) under nitrogen atmosphere. 1H and 13C NMR spectra were recorded using CDCl3 on Varian 300 MHz Ultrashield spectrometer with tetramethylsilane (TMS) as an internal reference at working frequency 300 MHz and 75 MHz, respectively. Fourier transform infrared (FT-IR) spectra were recorded on a Perkin Elmer Frontier 91,579. Mass spectrometric measurements were recorded using MALDI-TOF (Bruker) and elemental analysis was carried out on EA Euro-elemental analysis instrument. To confirm the formation of nanoparticles in AIE studies, the sample of synthesized dye was analysed by dynamic light scattering (DLS) technique using Zetasizer Ver. 7.12, serial number: MAL1180779.
Methylation of isatin and its derivatives
To increase the solubility of isatin and its –CH3, –Br, and –NO2 derivatives, N-methylation was done according to a reported procedure by Beauchard et al. [54].
General method for the synthesis of compounds 1–4
The synthetic method given by Bergman et al. [55] has been used for the preparation of 1–4, wherein a mixture of 3,4-diaminobenzophenone (1.0 mmol) and 5-substituted-1-methyl-1H-Indole-2,3-dione (1.2 mmol) were dissolved in 10 cm3 glacial acetic acid and refluxed for 3–4 h. The reaction mixture was cooled to room temperature and neutralized with sodium hydrogen carbonate to pH 7. The resulting solid obtained by vacuum filtration was washed thoroughly with H2O and kept for air-drying overnight. The crude product obtained was further purified with different ratios of n-hexane/chloroform by silica gel column chromatography to obtain a yellow to orange solid. (Compounds 1, 2, 3, and 4 were purified by using n-hexane/chloroform with ratio 90:10, 90:10, 80:20, and 70:30, respectively).
6-Methyl-6H-indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone (1, C22H15N3O)
Yellow solid; yield 286 mg (84%); m.p.: 261 °C; FT-IR (KBr): \(\overline{\nu }\) = 3050, 2919, 1710, 1650, 1583, 1257,1200, 752 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.82–8.03 (m, 4H), 8.01–7.79 (m, 2H), 7.76–7.30 (m, 6H), 3.94 (s, 3H, ‒NCH3) ppm; 13C NMR (75 MHz, CDCl3): δ = 196.27 (C=O), 145.47, 141.61, 141.07, 139.48, 137.09, 134.49, 132.54, 131.85, 137.10, 131.15, 130.17, 129.69, 128.83, 128.43, 125.93, 122.81, 121.81, 121.53, 121.33, 119.08, 109.39, 27.66 (‒NCH3) ppm; MALDI-TOF: m/z calcd for C22H15N3O 337.12 (M+), found 337.90.
6,9-Dimethyl-6H-indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone (2, C23H17N3O)
Yellow solid; yield 273 mg (81%); m.p.: 278 °C; FT-IR (KBr): \(\overline{\nu }\) = 3057, 2918, 2861, 1707, 1651, 1570, 1487, 1280, 1117, 803, 718 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.52–8.08 (m, 4H), 8.00–7.80 (m, 2H), 7.71–7.28 (m, 5H), 3.91 (s, 3H, ‒NCH3), 2.56 (s, 3H, ‒CCH3) ppm; 13C NMR (75 MHz, CDCl3): δ = 196.34 (C=O), 143.70, 141.05, 139.46, 136.96, 133.36, 133.05, 132.71, 132.52, 132.33, 131.14, 130.99, 130.17, 129.67, 128.53, 128.05, 125.86, 123.04, 122.70, 119.16, 109.12, 27.58 (‒NCH3), 21.29 (‒CCH3) ppm; MALDI-TOF: m/z calcd for C23H17N3O 351.41 (M+), found 351.97.
9-Bromo-6-methyl-6H-indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone (3, C22H14BrN3O)
Brown solid; yield 262 mg (78%); m.p.: 290 °C; FT-IR (KBr): \(\overline{\nu }\) = 3061, 2927, 2861, 1706, 1651, 1564, 1443, 1279, 1117, 803, 718 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.70–8.48 (m, 2H), 8.45–8.11 (m, 2H), 8.03–7.74 (m, 3H), 7.73–7.49 (m, 3H), 7.37 (d, J = 8.6 Hz, 1H), 3.95 (s, 3H, ‒NCH3) ppm; 13C NMR (75 MHz, CDCl3): δ = 196.06 (C=O), 143.94, 143.77, 140.35, 139.94, 134.09, 132.49, 132.14, 131.14, 130.19, 130.09, 130.06, 129.30, 128.58, 128.47, 128.31, 126.28, 126.24, 125.77, 122.70, 114.01, 110.98, 27.57 (‒NCH3) ppm; MALDI-TOF: m/z calcd for C22H14BrN3O 416.28 (M+), found 416.51 (M+), 418.43 ([M + 2]+).
6-Methyl-9-nitro-6H-indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone (4, C22H14N4O3)
Orange solid; yield 134 mg (40%); m.p.: > 300 °C; FT-IR (KBr): \(\overline{\nu }\) = 3063, 2918, 2849, 1719, 1651, 1582, 1502, 1487, 1320, 1117, 797, 709 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.72–8.47 (m, 2H), 8.42–8.27 (m, 1H), 8.25–8.12 (m, 1H), 8.05–7.77 (m, 3H), 7.70–7.46 (m, 3H), 7.43–7.33 (m, 1H), 3.97 (s, 3H, ‒NCH3) ppm; MALDI-TOF: m/z calcd for C22H14N4O3 382.11 (M+) found 382.28.
References
Friend RH, Gymer RW, Holmes AB, Burroughes JH, Marks RN, Taliani C, Bradley DDC, Dos Santos DA, Brédas JL, Lögdlund M, Salaneck WR (1999) Nature 397:121
Baldo MA, Thompson ME, Forrest SR (2000) Nature 403:750
Anthony JE (2008) Angew Chem Int Ed 47:452
Grätzel M (2009) Acc Chem Res 42:1788
Imahori H, Umeyama T, Ito S (2009) Acc Chem Res 42:1809
Ning Z, Tian H (2009) Chem Commun 7:5483
Staub K, Levina GA, Barlow S, Kowalczyk TC, Lackritz HS, Fort A, Marder SR (2003) J Mater Chem 13:825
Jiao GS, Thoresen LH, Burgess K (2003) J Am Chem Soc 1226:14668
Doré K, Dubus S, Ho HA, Lévesque I, Brunette M, Corbeil G, Boissinot M, Boivin G, Bergeron MG, Boudreau D, Leclerc M (2004) J Am Chem Soc 13:4240
Luo J, Xie Z, Lam JWY, Cheng L, Chen H, Qiu C, Kwok HS, Zhan X, Liu Y, Zhu D, Tang BZ (2001) Chem Commun 18:1740
Yuan WZ, Lu P, Chen S, Lam JWY, Wang Z, Liu Y, Kwok HS, Ma Y, Tang BZ (2010) Adv Mater 22:2159
Mei J, Leung NLC, Kwok RTK, Lam JWY, Tang BZ (2015) Chem Rev 115:11718
Zhang W, Yu G (2015) Organic optoelectronics materials. Springer, Switzerland, p 60
Zhang W, Yu G (2015) Organic optoelectronics materials. Springer, Switzerland, p 67
Quinn JTE, Zhu J, Li X, Wang J, Li Y (2017) J Mater Chem C 5:8654
Winkler M, Houk KN (2007) J Am Chem Soc 129:1805
Shaikh AM, Sharma BK, Chacko S, Kamble RM (2016) RSC Adv 6:60084
Shaikh AM, Sharma BK, Kamble RM (2015) J Chem Sci 127:1571
Kanekar DN, Chacko S, Kamble RM (2019) Dyes Pigm 167:36
Mahadik SS, Chacko S, Kamble RM (2019) ChemistrySelect 4:10021
Singh PS, Chacko S, Kamble RM (2019) N J Chem 43:6973
Sharma BK, Shaikh AM, Chacko S, Kamble RM (2017) J Chem Sci 129:483
Singh PS, Badani PM, Kamble RM (2019) N J Chem 43:19379
Shaikh AM, Sharma BK, Chacko S, Kamble RM (2016) RSC Adv 6:94218
Shaikh AM, Sharma BK, Chacko S, Kamble RM (2017) N J Chem 41:628
Kanekar DN, Chacko S, Kamble RM (2020) N J Chem 44:3278
Mahadik SS, Garud DR, Ware AP, Pingale SS, Kamble RM (2021) Dyes Pigm 184:108742
Singh PS, Ghadiyali M, Chacko S, Kamble RM (2022) J Lumin 242:118568
Kanekar DN, Chacko S, Kamble RM (2018) ChemistrySelect 3:4114
Sharma BK, Shaikh AM, Agarwal N, Kamble RM (2016) RSC Adv 6:17129
Shaikh AM, Chacko S, Kamble RM (2017) ChemistrySelect 2:7620
Harmenberg J, Wahren B, Bergman J, Akerfeldt S, Lundlad L (1988) Antimicrob Agents Chemother 32:1720
Harmenberg J, Akesson-Johansson A, Gräslund A, Malmfors T, Bergman J, Wahren B, Akerfeldt S, Lundblad L, Cox S (1991) Antiviral Res 15:193
Hirata K, Araya J, Nakaike S, Kitamura K, Ishida T (2001) Chem Pharm Bull 49:44
Deady LW, Kaye AJ, Finlay GJ, Baguley BC, Denny WA (1997) J Med Chem 40:2040
Manna K, Agrawal YK (2009) Bioorg Med Chem Lett 19:2688
Pai NR, Pusalkar DA (2010) J Chem Pharm Res 2:485
Sharma V, Kumar P, Pathaka D (2010) J Heterocycl Chem 47:491
Slater LS, Callis PR (1995) J Phys Chem 99:8572
Jennings P, Jones AC, Mount AR (1998) J Chem Soc Faraday Trans 94:3619
Walker MS, Bednar TW, Lumry R (1967) J Chem Phys 47:1020
Martinaud M, Kadiri A (1978) Chem Phys 28:473
Lakowicz JR (2006) Solvent and environmental effects. In: Lakowicz JR (ed) Principles of fluorescence spectroscopy. Springer, Boston
Zhu X, Su Q, Feng W, Li F (2017) Chem Soc Rev 46:1025
Kasai H, Nalwa HS, Oikawa H, Okada S, Matsuda H, Minami N, Kakuta A, Ono K, Mukoh A, Nakanishi H (1992) Jpn J Appl Phys 31:L1132
Yang W, Li C, Zhang M, Zhou W, Xue R, Liu H, Li Y (2016) Phys Chem Chem Phys 18:28052
Liu W, Ying S, Zhang Q, Ye S, Guo R, Ma D, Wang L (2018) Dyes Pigm 158:204
Li H, Chi Z, Zhang X, Xu B, Liu S, Zhang Y, Xu J (2011) Chem Commun 47:11273
Chikamatsu M, Itakura A, Yoshida Y, Azumi R, Yase K (2008) Chem Mater 20:7365
Byung B, Jung J, Lee K, Sun J, Andreou AG, Katz HE (2010) Adv Funct Mater 20:2930
Zheng Q, Huang J, Sarjeant A, Katz HE (2008) J Am Chem Soc 130:14410
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Cliford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C02. Gaussian Inc, Wallingford
Bottom R (2008) Thermogravimetric analysis. In: Gabbott P (ed) Principles and applications of thermal analysis. Wiley online books, Hoboken, p 87
Beauchard A, Ferandin Y, Frere S, Lozach O, Blairvacq M, Meijer L, Thiery V, Besson T (2006) Bioorg Med Chem 14:6434
Bergman J, Charlotta D (1996) Recl Trav Chim Pays-Bas 115:31
Acknowledgements
The authors are greatly thankful to Micro-Analytical Laboratory, Department of Chemistry, University of Mumbai for providing Instrumental facilities. We sincerely thank the Tata Institute of Fundamental Research (TIFR), Mumbai for providing MALDI-TOF. One of the author DNK is grateful to DST-PURSE for JRF.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kanekar, D.N., Dhanawade, S.S., Jadhav, A.B. et al. Fluorescent indolo[2,3-b]quinoxalin-2-yl(phenyl)methanone dyes: photophysical, AIE activity, electrochemical, and theoretical studies. Monatsh Chem 153, 895–906 (2022). https://doi.org/10.1007/s00706-022-02974-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-02974-0