
Abstract
Discovery of β-lactamase inhibitors is a continuous process due to inherent capability of resistance in bacteria against existing inhibitors. Diazabicyclooctane ring is a non-β-lactam motif capable of inhibiting the β-lactamases. As part of our efforts, we synthesized a series of diazabicyclooctane derivatives where C2 position of the bicyclic ring is linked with various substituted-amidine moieties. The newly synthesized compounds, alone and in combination with meropenem, were tested against ten bacterial strains for their antibacterial activity in vitro. All compounds did not show antibacterial tendency when tested alone (MIC > 64 mg/dm3) however, showed antibacterial activity in combination with meropenem. All compounds enhanced the potency of meropenem (MIC 2–4 mg/dm3) with MIC values ranging from < 0.125 to 1 mg/dm3 indicating their prospective β-lactamase inhibition capability. One derivative proved to be the most potent among all, and is comparable to avibactam, against eight out of ten bacterial strains.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In addition to other mechanisms, emergence of new β-lactamases is the major cause of antibiotic resistance in Gram-negative bacterial strains. β-Lactamase inhibition is an alternative strategy to cope with the antibiotic resistance where the inhibitor is employed in combination with existing antibiotic. With the prescription of multidrug-therapy a number of traditional β-lactam (BL) antibiotics restored their activity in combination with suitable β-lactamase inhibitors (BLI) such as clavulanic acid [1, 2], sulbactam, and tazobactam [3]. Clavulanic acid/amoxicillin (augmentin) combination successfully inactivated class A β-lactamases and extended spectrum β-lactamases (ESBLs) [4] for around 40 years of its first clinical application. Following, sulbactam and tazobactam combinations proved effective against class A, class C, as well as a few class D β-lactamases [5].
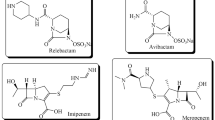
Since diazabicyclooctane (DBO) has been recognized as a synthetic alternative to BL ring [6], a series of second-generation inhibitors based on DBO functionalization have been prepared [7] and evaluated against various β-lactamases [8]. Worth mentioning are avibactam and relebactam which are potent inhibitors of class A carbapenemases (KPCs), AmpCs (class C), and some of class D lactamases [9]. These DBO based BLIs have been approved for clinical use against multiple bacterial indications in combination with existing BLs [10]. These multi-drugs are currently first line defence against extended spectrum β-lactamases (ESBLs), carbapenemases, MDR [11], Enterobacteriaceae as well as Pseudomonas aeruginosa [12,13,14]. However, these combinations are not effective against class B and most of class D β-lactamases. Therefore, several other DBO based BLIs (Fig. 1) [6, 15,16,17] are passing through phase I or phase III clinical trials [10, 18] in combination with different types of β-lactams (such as meropenem, cefepime, cefpodoxime, etc.).

Structures of diazabicyclooctane derivatives currently approved or in clinical trials
Recently we reported the synthesis and β-lactamase inhibition tendency of a number of amidine substituted DBOs [19,20,21,22], where NH2 group of amidine attached to the C2 of DBO ring was functionalized with numerous substituents ranging from alkyl to aryl and N-heterocyclics to N,S-heterocycles. Although all compounds showed variable lactamase inhibition capability, however, all compounds could not compete with avibactam. After careful examination of our previously generated data, we chose N-containing substituents to be explored further and here we report the synthesis and antibacterial activities of new compounds alone, as well as a combination with existing β-lactam, meropenem (MER).
Results and discussion
Synthesis of compounds 4a–4e is shown in Scheme 1. Synthesis of amidine compound 1 was accomplished by our previously reported method [19, 20] and it was obtained as diastereomeric mixture of R- and S-configuration. The S-isomer was found to be antibacterial as compared to its counterpart [19, 20, 23].

Reagents and conditions: (i) HO-Ra-e, HATU, DIPEA, DMF/CH2Cl2, RT, 16 h, 44-94%; (ii) Pd/C (wet), THF/TEA, H2, RT, 16 h, 36%-quantitative; (iii) SO3-pyridine or SO3-NMe3, pyridine or TEA, RT, 16 h, Dowex-50wx Na+, 2-70%
The phenomenon of racemization at this stage was surveyed in literature and it was found that the racemization at C2 of DBO has been observed while reacting similar substrate with ammonia [23]. Therefore, based on our own observations and literature reports, we propose the following mechanism for this epimerization (Scheme 2). We have observed the evolution of a gas during the addition of AlMe3 into NH4Cl suspension, indicating the formation of the methyl aluminum amide [24, 25] which acts as source of ammonia and a mild base. Hydrogen abstraction at C2 of the compound A generates the enimine intermediate B, which upon nucleophilic addition of ammonia converts to regioisomers through the formation of intermediate C (Scheme 2).

Next step was the coupling of the organic acids with amidine 1 to form the corresponding derivatives 2a–2e. Classical coupling reagent such as HATU [26] (O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate) was used in DMF (dimethylformamide) or CH2Cl2 in the presence of DIPEA (N,N-diisopropylethylamine) as a base. For the preparation of 2c, (S)-1-methylpiperidine-3-carboxylic acid was required which was prepared from (S)-piperidine-3-carboxylic acid by slight modification to a known procedure [27]. Thus, (S)-piperidine-3-carboxylic acid dissolved in methanol was reacted with paraformaldehyde in the presence of palladium catalyst under hydrogen atmosphere at 50 °C for 16 h. Similarly (R)-1-methylpyrrolidine-3-carboxylic acid was prepared [27] from (R)-pyrrolidine-3-carboxylic acid before its condensation with compound 1 to yield the compound 2d. Palladium catalysed hydrogenation of compounds 2a–2e in THF led to the debenzylation affording hydroxy derivatives 3a–3e. Compounds 3a–3e were then reacted with SO3-pyridine complex or SO3-NMe3 to form sulfonic acid derivatives 4a–4e after purification by preparative HPLC. Sodium salts of these compounds were obtained by ion exchange using column filled with Dowex-50wx Na+ resin. Water was used as an eluant which was removed by lyophilization to obtain the sodium salts of desired compounds (Scheme 1).
Synthesis of compound 4f was accomplished following the Scheme 3. Amidine 1 was condensed with 3-[(tert-butoxycarbonyl)amino]propanoic acid using DCC (N,N′-dicyclohexylcarbodiimide) and DMAP (4-dimethylaminopyridine) as the coupling reagents to yield 90% of compound 5. Benzyl deprotection of 5 was achieved by Pd/C catalysed hydrogenation in EtOAc. It has been observed that a catalytic amount of triethylamine in EtOAc enhances the rate of hydrogenolysis of benzyl ether derivatives [28]. Final compound 4f was obtained by sulfonation of 6 followed by deprotection of Boc group by reacting the intermediate sulfonic acid derivative with TFA (trifluoroacetic acid) in CH2Cl2 at low temperature and subsequent ion exchange by Dowex-50wx Na+ resin.

Reagents and conditions: (i) 3-[(tert-butoxycarbonyl)amino]propanoic acid, DCC, DMAP, THF, RT, 16 h, 90%; (ii) Pd/C (wet), EtOAc/TEA, H2, RT, 16 h, 69%; (iii) SO3-pyridine, pyridine, RT, 16 h, 94% (crude); (iv) TFA, CH2Cl2, 0 °C, 3.5 h, Dowex-50wx Na+, 10%
Synthesis of analogous compound 4g was accomplished following the procedures elaborated in Scheme 4 because the benzyl deprotection was unsuccessful in this case while following the Scheme 3. Therefore, we prepared TBS (tert-butyldimethylsilyl) protected amidine compound 7 following our previously described method [19, 20]. Compound 7 was then successfully coupled with 2-(1H-imidazol-1-yl)acetic acid using HATU in DMF/CH2Cl2 mixture as solvent in the presence of DIPEA. TBS protection in compound 8 was then removed by TBAF (tetrabutylammonium fluoride) leading to the formation of hydroxy derivative 9, which was converted to the sodium salt of 4g using the similar procedure described for 4a–4e. All the final compounds 4a–4g were purified by preparative HPLC, and the structures were confirmed by NMR spectra as well as HRMS data.

Reagents and conditions: (i) 2-(1H-imidazol-1-yl)acetic acid, HATU, DIPEA, DMF/CH2Cl2, RT, 24 h, 57%; (ii) TBAF, THF, 0 °C, 1.5 h, 98%; (iii) SO3-NMe3, TEA, THF/H2O, Dowex-50wx Na+, 44%
Substituted-amidine derivatives 4a–4g were then analysed for their antibacterial activity in vitro alone, as well as in combination with meropenem, an existing antibiotic, using broth dilution method [29]. A number of bacterial species with different β-lactamases (Table 1), i.e. E. coli clinical isolate; E. coli 8739; K. pneumoniae clinical isolate; K. pneumoniae 700603; E. cloacae clinical isolate; E. cloacae 700323; A. baumannii clinical isolate; A. baumannii 19606; P. aeruginosa clinical isolate, and P. aeruginosa 9027, were used as test bacterial species. Meropenem (MER) alone, and as a combination with avibactam, as well as with synthesized compounds 4a–4g, were analysed for antimicrobial activity by determining their minimum inhibitory concentration (MIC, mg/dm3) values. For comparison MIC values of avibactam and newly synthesized compounds 4a–4g without meropenem were also determined.
It was observed that the compounds 4a–4g are not antibacterial in action (MIC > 64 mg/dm3) when used alone without β-lactam, but antibacterial activity of the β-lactam meropenem was changed upon combining the compounds 4a–4g individually (Table 1). This implies the formation of stable complex of the derivatives 4a–4g with β-lactamases present in the bacterial strains, thus indicating the β-lactamase inhibitor potential of the synthesized compounds. In general, all compounds 4a–4g enhanced potency of meropenem against all tested species with MIC values ranging from < 0.125 to 1 mg/dm3 (compared to MER, MIC 2–4 mg/dm3).
To our delight all compounds showed potency comparable to avibactam in combination with meropenem (MER + Avibactam), against E. coli clinical isolate, E. coli 8739, K. pneumoniae clinical isolate, K. pneumoniae 700603, A. baumannii clinical isolate, and A. baumannii 19606. All compounds except 4b showed similar or better activity against P. aeruginosa clinical isolate (MIC 0.25–0.5 mg/dm3) compared to avibactam (MIC 0.5 mg/dm3). Among all bacterial species, E. cloacae strains proved the most resistant against all compounds with MIC values of 0.25 mg/dm3 (for avibactam MIC is < 0.125 mg/dm3), except for compound 4g where MIC was comparable to avibactam against E. cloacae 700323 (MIC < 0.125 mg/dm3).
It is worth mentioning that the stereochemistry of the final compounds does impact the antibacterial strength. This can be assessed by the activity comparison of the stereochemical isomers of compounds 4c and 4d previously reported by us. The compounds 4c and 4d are comparatively more active against all these bacterial strains in vitro [20]. From the data in Table 1, it is evident that compounds 4b and 4c showed better activity than avibactam against A. baumannii 19606, whereas compounds 4d, 4e, and 4f were better than avibactam against P. aeruginosa clinical isolate. Moreover, it is clear that the compound 4g is the most potent among all compounds and showed comparable potency to avibactam against eight out of ten strains and even better potency against one species.
Conclusion
We successfully synthesized seven substituted-amidine derivatives of diazabicyclooctane and the compounds were tested for antibacterial activity alone, and in combination with meropenem using bacterial species containing different β-lactamases. All compounds were not antibacterial in nature when tested alone, however all the compounds enhance the antibacterial strength of the antibiotic in vitro. This indicates the β-lactamase inhibition capability of the compounds allowing the drug to kill the bacteria, through a synergism between antibiotic and lactamase inhibitor. The compound 4g proved to be the most potent among all and may be a lead compound for further development, and preclinical studies.
Experimental
All 1H and 13C NMR spectra were recorded on a Bruker AVANCE NEO 400 NMR operating at 400 MHz for 1H and 100 MHz for 13C, respectively (see supplementary figures). The NMR data were recorded either in D2O or DMSO-d6 containing 0.03% TMS as internal standard. Chemical shifts (δ) are given in parts per million (ppm) relative to TMS and coupling constant (J values) are given in Hertz (Hz). Signal multiplicities are reported as follows: s, singlet; brs, broad singlet; d, doublet; t, triplet and m, multiplet. IR spectra were measured on a SHIMADZU IRSpirit Fourier Transform Infrared Spectrophotometer. Melting points were measured on SGW X-4 (Laboratory device). Preparative HPLC was performed on an Agilent 1260 Infinity II System on Agilent 10 prep-C18 250 × 21.2 mm column, using an acetonitrile/aqueous 0.1% trifluoroacetic acid gradient, or an acetonitrile/aqueous 0.1% formic acid gradient, or an acetonitrile/water at 22 °C. Mass spectra were performed on an Agilent 1260II-6125 Separation Module using either ES− or ES+ ionization modes. Column Chromatography was performed with using Qingdao Inc. Silica Gel: CC Grade (230-400 Mesh). Commercial solvents and reagents were generally used without further purification. All products were dried before characterization and used in subsequent synthetic steps.
General method for the synthesis of compounds 2a-2e; N-[[(2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]nicotinamide (2a, C20H21N5O3)
To a solution of 500 mg 1 (1.82 mmol), 1.04g HATU (2.73 mmol), and 707 mg DIPEA (5.47 mmol) dissolved in 5 cm3 DMF, 337 mg nicotinic acid (2.73 mmol) was added at room temperature under N2. The reaction mixture was stirred for 16 h and was diluted with CH2Cl2 upon completion of the reaction. The organic phase was separated, and washed with brine and saturated NH4Cl, dried over Na2SO4, and filtered. The organic layer was concentrated to give a residue, which was purified by silica gel column chromatography eluting with 50% EtOAc in hexane to give the title compound 2a as a white solid, which was directly used for next step without further purification. Yield 75%; MS (LC/MS): m/z = 380.4 ([M + H]+).
N-[[(2S,5R)-6-(Benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]isonicotinamide (2b, C20H21N5O3)
Compound 2b was prepared from 337 mg isonicotinic acid (2.73 mmol) and 500 mg 1 (1.82 mmol) according to the general method. Yield 650 mg (94%) as a white solid; MS (LC/MS): m/z = 380.4 ([M + H]+).
(3S)-N-[[(2S,5R)-6-(Benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-1-methylpiperidine-3-carboxamide (2c, C21H29N5O3)
215 mg (S)-1-methylpiperidine-3-carboxylic acid (1.50 mmol) and 215 mg 1 (0.75 mmol) were reacted in 5 cm3 CH2Cl2/DMF (3:2) for 30 h according to the general procedure. Yield 195 mg (65%); 1H NMR (400 MHz, DMSO-d6): δ = 1.54 (brs, 4H), 1.65–1.74 (m, 2H), 1.88–1.97 (m, 2H), 2.35 (brs, 3H), 2.78 (brs, 3H), 2.91 (brs, 1H), 3.04 (brs, 1H), 3.19 (brs, 1H), 3.35 (brs, 1H), 3.80–3.91 (m, 2H), 4.85 (brs, 2H), 5.66 (s, 1H), 6.76 (s, 2H), 7.36–7.39 (m, 3H), 7.49–7.51 (m, 2H) ppm; MS (LC/MS): m/z = 400.2 ([M + H]+).
(3R)-N-[[(2S,5R)-6-(Benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-1-methylpyrrolidine-3-carboxamide (2d, C20H27N5O3)
Compound 2d was synthesized from 330 mg 1 (1.20 mmol) and 300 mg (R)-1-methylpyrrolidine-3-carboxylic acid (2.33 mmol) in 6 cm3 CH2Cl2/DMF (1:1) for 48 h, according to general method. Yield 204 mg (44%) brown solid; 1H NMR (400 MHz, DMSO-d6): δ = 1.72–1.79 (m, 1H), 1.90–2.03 (m, 5H), 2.22–2.27 (m, 1H), 2.60 (s, 3H), 2.95–2.97 (m, 1H), 3.16–3.27 (m, 3H), 3.48–3.51 (m, 1H), 3.82–3.87 (m, 1H), 3.90–3.97 (m, 1H), 4.85 (brs, 2H), 5.68 (s, 1H), 6.74 (s, 2H), 7.35–7.41 (m, 3H), 7.47–7.53 (m, 2H) ppm; MS (LC/MS): m/z = 386.2 ([M + H]+).
N-[[(2S,5R)-6-(Benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-2-(piperidin-1-yl)acetamide (2e, C21H29N5O3)
Compound 2e was prepared from 260 mg 2-(piperidin-1-yl)acetic acid (1.45 mmol) and 230 mg 1 (0.84 mmol) according to the general method. Yield 245 mg (73%) yellow solid; 1H NMR (400 MHz, DMSO-d6): δ = 1.33–1.74 (m, 7H), 1.78–2.08 (m, 4H), 2.22–2.43 (m, 2H), 2.78 (t, J = 12.1 Hz, 1H), 3.22–3.32 (m, 3H), 3.75–3.85 (m, 1H), 3.94–4.09 (m, 1H), 4.34 (t, J = 14.3 Hz, 1H), 4.83–4.89 (m, 2H), 5.64 (s, 1H), 6.73 (s, 2H), 7.34–7.41 (m, 3H), 7.46–7.53 (m, 2H) ppm; MS (LC/MS): m/z = 400.2 ([M + H]+).
General method for the synthesis of compounds 3a-3e; N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]nicotinamide (3a, C13H15N5O3)
100 mg of 10% wet Pd/C was added to a solution of 200 mg compound 2a (0.52 mmol) dissolved in 3 cm3 THF containing few drops of triethylamine. The mixture was stirred under H2 atmosphere at room temperature for 16 h. Later, the reaction mixture was filtered through a pad of celite, rinsed with EtOAc, and the filtrate was concentrated in vacuum. The crude product was purified by flash silica gel chromatography using 10% MeOH in CH2Cl2 to give the title compound 3a (67 mg, 44% in two steps) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ = 1.75–1.83 (m, 1H), 1.87–2.09 (m, 3H), 3.13–3.24 (m, 1H), 3.45–3.62 (m, 1H), 4.07–4.15 (m, 1H), 5.70 (s, 1H), 6.44 (s, 2H), 7.51 (dd, J = 7.8, 4.8 Hz, 1H), 7.93 (dt, J = 7.8, 1.7 Hz, 1H), 8.67–8.71 (m, 2H), 9.24 (s, 1H) ppm; MS (LC/MS): m/z = 290.2 ([M + H]+).
N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]isonicotinamide (3b, C13H15N5O3)
Compound 3b was synthesized from 650 mg 2b (1.71 mmol) using general method. Yield 36% white solid; 1H NMR (400 MHz, DMSO-d6): δ = 1.75–1.82 (m, 1H), 1.87–2.11 (m, 3H), 3.16–3.25 (m, 1H), 3.33–3.43 (m, 1H), 4.04–4.13 (m, 1H), 5.76 (s, 1H), 6.43 (s, 2H), 7.47 (d, J = 4.9 Hz, 2H), 8.70 (d, J = 4.9 Hz, 2H), 9.20 (s, 1H) ppm; MS (LC/MS): m/z = 290.2 ([M + H]+).
(3S)-N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-1-methylpiperidine-3-carboxamide (3c, C14H23N5O3)
Compound 3c was synthesized according to general procedure from 195 mg compound 2c (0.49 mmol). Yield 155 mg (quantitative) white solid, which was directly used for next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ = 1.46–1.79 (m, 4H), 1.83–2.04 (m, 4H), 2.21 (s, 3H), 2.70–2.97 (m, 5H), 3.16–3.26 (m 1H), 3.73–3.82 (m, 1H), 3.87–3.98 (m, 1H), 5.65 (s, 1H), 6.46 (s, 2H), 9.28 (s, 1H) ppm; MS (LC/MS): m/z = 310.2 ([M + H]+).
(3R)-N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-1-methylpyrrolidine-3-carboxamide (3d, C13H21N5O3)
Compound 3d was prepared form 200 mg 2d (0.52 mmol) according to the general procedure. Yield 160 mg (quantitative) white solid. Compound 3d was directly used for next step without further purification. MS (LC/MS): m/z = 296.2 ([M + H]+).
N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-2-(piperidin-1-yl)acetamide (3e, C14H23N5O3)
240 mg compound 2e (0.60 mmol) was hydrogenated according to the general procedure to get the 190 mg compound 3e. Yield quantitative, white solid. Compound 3e was directly used for next step without further purification. MS (LC/MS): m/z = 310.2 ([M + H]+).
General method for the synthesis of compounds 4a-4e; Sodium (2S,5R)-2-(N-nicotinoylcarbamimidoyl)-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4a, C13H14N5NaO6S)
A mixture of 406 mg compound 3a (1.45 mmol) and 569 mg SO3-pyridine (7.27 mmol) dissolved in 20 cm3 pyridine was stirred at room temperature for 16 h. The reaction mixture was concentrated to give a residue, which was purified by preparative HPLC on an Agilent 10 prep-C18 250 × 21.2 mm column and lyophilized, followed by ion exchange using column filled with Dowex-50wx Na+ resin, to give 4a. Yield 12 mg (2%) white solid; m.p.: 182.3 °C; 1H NMR (400 MHz, D2O): δ = 1.85–2.09 (m, 2H), 2.11–2.23 (m, 2H), 3.50–3.61 (m, 1H), 3.77–2.86 (m, 1H), 4.04–4.13 (m, 1H), 5.75 (s, 1H), 7.53 (dd, J = 5.0, 7.8 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 8.59 (d, J = 1.5 Hz, 1H), 8.62 (d, J = 5.0 Hz, 1H) ppm; 13C NMR (100 MHz, D2O): δ = 26.5, 27.8, 41.6, 57.2, 61.1, 116.9, 124.1, 129.4, 136.2, 146.8, 150.7, 162.6, 170.1 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3487, 3355, 2963, 2250, 1592, 1058, 856, 741, 597 cm−1; MS (LC/MS): m/z = 368.1 ([M-Na]−); MS (HR-EI-MS): m/z = 368.0660.
Sodium (2S,5R)-2-(N-isonicotinoylcarbamimidoyl)-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4b, C13H14N5NaO6S)
200 mg compound 3b (0.72 mmol) was reacted with 569 mg SO3-pyridine (3.58 mmol) as described in general method to afford compound 4b. Yield 5 mg (2%) white solid; m.p.: 190.1 °C; 1H NMR (400 MHz, D2O): = δ 1.92–2.07 (m, 2H), 2.10–2.23 (m, 2H), 3.49–3.58 (m, 1H), 3.67–3.75 (m, 1H), 4.01–4.10 (m, 1H), 5.76 (s, 1H), 7.46 (d, J = 5.0 Hz, 2H), 8.61 (d, J = 4.7 Hz, 2H) ppm; 13C NMR (100 MHz, D2O): = δ 26.5, 27.6, 41.4, 58.8, 57.1, 116.8, 121.0, 121.6, 141.8, 149.2, 149.5, 162.5, 169.7 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3475, 3392, 2965, 1691, 1648, 1058, 845, 741, 600 cm−1; MS (LC/MS): m/z = 368.1 ([M-Na]−); MS (HR-EI-MS): m/z = 368.0659.
Sodium (2S,5R)-2-[N-[(S)-1-methylpiperidine-3-carbonyl]carbamimidoyl]-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4c, C14H22N5NaO6S)
Compound 4c was synthesized from 155 mg 3c (0.5 mmol) as described in general procedure. Yield 134 mg (70%) white solid; m.p.: 184 °C; 1H NMR (400 MHz, D2O): δ = 1.75–2.03 (m, 6H), 2.08–2.18 (m, 2H), 2.82 (s, 3H), 2.86–2.95 (m, 1H), 3.02–3.11 (m, 1H), 3.37–3.67 (m, 4H), 3.95–4.08 (m, 2H), 5.70 (s, 1H) ppm; 13C NMR (100 MHz, D2O): δ = 23.8, 26.3, 34.0, 37.1, 40.7, 41.0, 43.4, 53.9, 54.0, 54.3, 54.5, 117.0, 162.7, 173.7 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3467, 3378, 2963, 1695, 1648, 1053, 972, 799, 560 cm−1; MS (LC/MS): m/z = 388.2 ([M-Na]−); MS (HR-EI-MS): m/z = 388.1296.
Sodium (2S,5R)-2-[N-[(R)-1-methylpyrrolidine-3-carbonyl]carbamimidoyl]-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4d, C13H20N5NaO6S)
A mixture of 160 mg compound 3d (0.54 mmol), 147 mg SO3-NMe3 (1.04 mmol), and 0.22 cm3 TEA (1.55 mmol) in 6 cm3 THF/water (1:1) was stirred at room temperature for 21 h. The reaction mixture was concentrated under reduced pressure to provide a residue. The residue was further purified by Dowex-50wx Na+ resin, using water as an elution solvent to give 4d. Yield 75 mg (38%) white solid; m.p.: 170.2 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.75–1.85 (m, 1H), 1.96–2.06 (m, 2H), 2.07–2.23 (m, 3H), 2.58–2.68 (m, 1H), 2.90 (s, 3H), 3.54–3.62 (m, 1H), 3.64–3.71 (m, 2H), 3.75–3.83 (m, 1H), 3.87–4.08 (m, 3H), 5.66 (s, 1H) ppm; 13C NMR (100 MHz, DMSO-d6): = δ 23.7, 27.2, 28.0, 41.2, 45.4, 47.6, 55.2, 55.7, 55.8, 56.9, 118.0, 161.5, 171.2 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3466, 3370, 2967, 1695, 1654, 1052, 972, 799, 621 cm−1; MS (LC/MS): m/z = 374.1 ([M-Na]−); MS (HR-EI-MS): m/z = 374.1138.
Sodium (2S,5R)-7-oxo-2-[N-[2-(piperidin-1-yl)acetyl]carbamimidoyl]-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4e, C14H22N5NaO6S)
Compound 4e was obtained from 190 mg 3e (0.6 mmol) using the procedure described for compound 4d. Yield 65 mg (65%) white solid; m.p.: 172.1 °C; 1H NMR (400 MHz, D2O): δ = 1.67–1.87 (m, 6H), 1.90–2.01 (m, 1H), 2.04–2.23 (m, 3H), 3.25–3.38 (m, 2H), 3.48–3.60 (m, 2H), 3.71–3.78 (m, 1H), 3.97–4.05 (m, 1H), 4.13–4.25 (m, 2H), 4.35 (t, J = 7.1 Hz, 1H), 5.63 (s, 1H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 21.9, 22.8, 23.2, 27.2, 28.5, 41.1, 45.2, 54.0, 55.1, 117.9, 161.5, 163.4 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3447, 2958, 1671, 1293, 1055, 972, 799, 621, 598 cm−1; MS (LC/MS): m/z = 388.1 ([M-Na]−); MS (HR-EI-MS): m/z = 388.1298.
tert-Butyl [3-[(2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboximidamido]-3-oxopropyl]carbamate (5, C22H31N5O5)
To a solution of 301 mg 1 (1.10 mmol), 70 mg DMAP (0.57 mmol), and 358 mg DCC (1.73 mmol) dissolved in 40 cm3 THF, 335 mg of 3-[(tert-butoxycarbonyl)amino]propanoic acid (1.77 mmol) was added at room temperature. The reaction mixture was then stirred for 16 h under nitrogen. After completion, the reaction mixture was concentrated to dryness, diluted with EtOAc, washed with saturated NH4Cl, brine, dried over Na2SO4, and filtered. The organic layer was concentrated to give a residue, which was purified by silica gel column chromatography eluting with 50–75% EtOAc in petroleum ether to give the title compound 5. Yield 440 mg (90%) white solid; 1H NMR (400 MHz, DMSO-d6): δ = 1.37 (s, 9H), 1.68–1.75 (m, 1H), 1.80–2.00 (m, 3H), 2.43–2.51 (m, 2H), 3.09–3.17 (m, 2H), 3.20–3.25 (m, 1H), 3.72–3.81 (m, 1H), 3.89–3.98 (m, 1H), 4.84 (s, 2H), 5.65 (s, 1H), 6.72 (s, 3H), 7.35–7.41 (m, 3H), 7.47–7.52 (m, 2H) ppm; MS (LC/MS): m/z = 446.2 ([M + H]+).
tert-Butyl [3-[(2S,5R)-6-hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboximidamido]-3-oxopropyl]carbamate (6, C15H25N5O5)
Hydrogenolysis of 440 mg compound 5 (0.99 mmol) was performed by the method described for 3a in 20 cm3 EtOAc with a few drops of TEA. The mixture was hydrogenated under 45 psi pressure at room temperature for 1 h, filtered through a pad of celite, rinsed with EtOAc. The filtrate was concentrated to give a residue, which was purified by silica gel column chromatography eluting with 5% MeOH in CH2Cl2 to give the title compound 6. Yield 245 mg (69%) yellow solid; 1H NMR (400 MHz, DMSO-d6): δ = 1.37 (s, 9H), 1.67–1.77 (m, 2H), 1.83–1.91 (m, 1H), 1.94–2.01 (m, 1H), 2.44–2.51 (m, 2H), 3.09–3.21 (m, 3H), 3.66–3.72 (m, 1H), 3.93–4.03 (m, 1H), 5.65 (s, 1H), 6.45 (s, 2H), 6.72 (t, J = 5.1 Hz, 1H), 9.27 (s, 1H) ppm; MS (LC/MS): m/z = 378.2 ([M + Na]+).
Sodium (2S,5R)-2-[N-(3-aminopropanoyl)carbamimidoyl]-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4f, C10H16N5NaO6S)
Compound 4f was prepared according to modified procedure described for 4a. 245 mg compound 6 (0.69 mmol) was reacted with 159 mg SO3-pyridine (1 mmol) in 10 cm3 pyridine, to form the intermediate sulfonic acid (332 mg, 94%) as a white foam. The intermediate was then treated with 1.4 cm3 TFA in 6 cm3 anhydrous CH2Cl2. The mixture was stirred for 3.5 h at 0 ºC, and then concentrated under reduced pressure to give a residue. The residue was dissolved in 15 cm3 CH2Cl2 and extracted with water (2 × 10 cm3). The aqueous layer was freeze-dried and purified by Dowex-50wx Na+ resin, using water as an elution solvent. Yield 22 mg (10%) white powder; m.p.: 203.5 °C; 1H NMR (400 MHz, D2O): δ = 1.71–1.80 (m, 1H), 1.89–1.97 (m, 1H), 2.02–2.11 (m, 2H), 2.77–3.87 (m, 2H), 3.17 (t, J = 6.0 Hz, 2H), 3.46–3.54 (m, 1H), 3.91–4.00 (m, 2H), 5.64 (d, J = 4.6 Hz, 1H) ppm; 13C NMR (100 MHz, D2O): δ = 22.6, 26.5, 29.9, 35.2, 40.9, 45.0, 57.4, 117.5, 162.8, 171.1 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3462, 3174, 2974, 1468, 1447, 1244, 1053, 972, 742, 598 cm−1; MS (LC/MS): m/z = 334.1 ([M-Na]−); MS (HR-EI-MS): m/z = 334.0829.
N-[[(2S,5R)-6-(tert-Butyldimethylsilyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-2-(1H-imidazol-1-yl)acetamide (8, C18H30N6O3Si)
704 mg HATU (1.85 mmol) and 0.82 cm3 DIPEA (4.08 mmol) were added to a solution of 490 mg 2-(1H-imidazol-1-yl)acetic acid (1.87 mmol) and 279 mg amidine 7 [19] (0.94 mmol) in 7 cm3 DMF/CH2Cl2 (1:1). The reaction mixture was then stirred at room temperature for 24 h. The reaction was quenched with saturated aq. NaHCO3 and extracted with EtOAc. The organic layer was washed with water, then brine, dried over Na2SO4 and filtered. The filtrate was concentrated to give a residue, which was purified by silica gel column chromatography eluting with 5% MeOH in CH2Cl2 containing 0.2% TEA to give the title compound 8. Yield 220 mg (57%) yellow solid. MS (LC/MS): m/z = 407.2 ([M + H]+).
N-[[(2S,5R)-6-Hydroxy-7-oxo-1,6-diazabicyclo[3.2.1]octan-2-yl](imino)methyl]-2-(1H-imidazol-1-yl)acetamide (9, C12H16N6O3)
0.65 cm3 TBAF (0.65 mmol) was added to a solution of 220 mg compound 8 (0.54 mmol) dissolved in 5 cm3 THF at 0 °C, and stirred at 0 °C for 1.5 h. The mixture was concentrated to dryness, added 5 cm3 of water, extracted with EtOAc and DCM. The organic layer was dried over Na2SO4, and filtrated. The filtrate was concentrated to give the title compound 9. Yield 188 mg (98%) brown solid, which was directly used for next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ = 1.68–1.78 (m, 2H), 1.90–2.05 (m, 2H), 3.19–3.25 (m, 1H), 3.68–3.77 (m, 1H), 4.05–4.13 (m, 1H), 5.08 (d, J = 17.1 Hz, 1H)), 5.14 (d, J = 17.1 Hz, 1H), 5.60 (s, 1H), 6.32 (s, 2H), 6.85 (s, 1H), 7.06 (s, 1H), 7.53 (s, 1H) ppm; MS (LC/MS): m/z = 293.1 ([M + H]+).
Sodium (2S,5R)-2-[N-[2-(1H-imidazol-1-yl)acetyl]carbamimidoyl]-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfate (4g, C12H15N6NaO6S)
Compound 4g was prepared from 188 mg compound 9 (0.64 mmol), 177 mg SO3-NMe3 (1.27 mmol), and 0.25 cm3 TEA (1.79 mmol) in 6 cm3 THF/water (1:1) as described for compound 4d. Yield 95 mg (44%) white solid; m.p.: 180.2 °C; 1H NMR (400 MHz, D2O): δ = 1.77–1.89 (m, 1H), 1.95–2.03 (m, 1H), 2.07–2.17 (m, 2H), 3.60–3.67 (m, 1H), 3.92–3.98 (m, 1H), 4.05–4.14 (m, 1H), 5.22 (d, J = 17.4 Hz, 1H), 5.29 (d, J = 17.4 Hz, 1H), 5.63 (d, J = 5.3 Hz, 1H), 7.20–7.23 (m 2H), 8.17 (s, 1H) ppm; 13C NMR (100 MHz, D2O): δ = 22.3, 26.3, 41.3, 44.4, 48.8, 57.0, 116.7, 122.0, 123.6, 137.5, 162.7, 167.5 ppm; FT-IR (KBr): \(\overline{\nu }\) = 3467, 3376, 3151, 2970, 1679, 1480, 1463, 1053, 972, 799, 613, 560 cm−1; MS (LC/MS): m/z = 371.1 ([M-Na]−); MS (HR-EI-MS): m/z = 371.0774.
Determination of antibacterial activity
Five wild type bacterial strains, namely E. coli clinical isolate (TEM-1), E. coli 8739 (CTX-M15), K. pneumoniae 700603 (KPC-3, TEM-1), E. cloacae 700323 (AmpC), A. baumannii 19606 (OXA-24), and P. aeruginosa 9027 (AmpC), containing different β-lactamases (indicated in parenthesis) were purchased from China Pharmaceutical Culture Collection (CPCC), whereas five species of clinical isolates such as E. coli clinical isolate (TEM-1), K. pneumoniae clinical isolate (SHV-1), E. cloacae clinical isolate (P99), A. baumannii clinical isolate (OXA-23/40) and P. aeruginosa clinical isolate (KPC-2) expressing variable β-lactamases were indigenously isolated and cultured from patients at Ningxia Medical University hospital, P.R. China.
A standard procedure was employed for these studies using the broth microdilution method according to the guidelines of the Clinical Laboratories and Standards Institute [29]. In a typical experiment, meropenem as a test antibiotic compound was dissolved in DMSO and diluted in microbial growth medium (Mueller–Hinton Broth II, cation adjusted) to a final concentration range of 0.125–64 mg/dm3 in serial two-fold dilution. In all cases the final DMSO concentration was less than 0.5%. Bacteria were added to 96-well microtiter plates containing the serial two-fold dilutions of the compounds; the final cell density was approximately 5 × 105 colony forming units/cm3 (CFU/cm3). Plates were incubated at 37 ºC for 18–24 h and read visually. The MIC of the test compound that inhibited visible growth of the bacteria was recorded. The same assay conditions were used when the compounds 4a–4g and avibactam alone and as a combination with test meropenem as an antibiotic compound (as two controls) were tested for MIC (mg/dm3). While meropenem was serially diluted as described above, a constant concentration (4 mg/dm3) of inhibitor compounds 4a–4g was used. The test results listed in Table 1.
References
Saudagar PS, Survase SA, Singhal RS (2008) Biotechnol Adv 26:335
Finlay J, Miller L, Poupard JA (2003) J Antimicrob Chemother 52:18
Shlaes DM (2013) Ann NY Acad Sci 1277:105
Ball P (2007) Int J Antimicrob Agents 30(Suppl 2):S113
Bush K, Bradford PA (2016) Cold Spring Harb Perspect Med 6:a025247
González-Bello C, Rodríguez D, Pernas M, Rodríguez A, Colchón E (2020) J Med Chem 63:1859
Deketelaere S, Van Nguyen T, Stevens CV, D’Hooghe M (2017) ChemistryOpen 6:301
Bouchet F, Atze H, Fonvielle M, Edoo Z, Arthur M, Ethève-Quelquejeu M, Iannazzo L (2020) J Med Chem 63:5257
Papp-Wallace KM, Nguyen NQ, Jacobs MR, Bethel CR, Barnes MD, Kumar V, Bajaksouzian S, Rudin SD, Rather PN, Bhavsar S, Ravikumar T, Deshpande PK, Patil V, Yeole R, Bhagwat SS, Patel MV, van den Akker F, Bonomo RA (2018) J Med Chem 61:4067
Butler MS, Paterson DL (2020) J Antibiot 73:329
Köck R, Cuny C (2020) Med Klin. Intensivmed Notfallmed 115:189
Nichols WW, Newell P, Critchley IA, Riccobene T, Das S (2018) Antimicrob Agents Chemother 62:e02446
Duin D, Bonomo RA (2016) Clin Infect Dis 63:234
Rodriguez BA, Girotto JE, Nicolau DP (2018) Curr Pediatr Rev 14:97
Morinaka A, Tsutsumi Y, Yamada M, Suzuki K, Watanabe T, Abe T, Furuuchi T, Inamura S, Sakamaki Y, Mitsuhashi N, Ida T, Livermore DM (2015) J Antimicrob Chemother 70:2779
Gordon EM, Duncton MAJ, Gallop MA (2018) J Med Chem 61:10340
Mushtaq S, Vickers A, Woodford N, Livermore DM (2017) J Antimicrob Chemother 72:1688
Tehrani K, Martin NI (2018) MedChemComm 9:1439
Iqbal Z, Zhai L, Gao Y, Tang D, Ma X, Ji J, Sun J, Ji J, Liu Y, Jiang R, Mu Y, He L, Yang H, Yang Z (2021) Beilstein J Org Chem 17:711
Gao Y, Liu Y, Iqbal Z, Sun J, Ji J, Zhai L, Tang D, Ji J, He L, Mu Y, Yang H, Yang Z (2021) ChemistrySelect 6:1174
Sun J, He L, Gao Y, Zhai L, Ji J, Liu Y, Ji J, Ma X, Mu Y, Tang D, Yang H, Iqbal Z, Yang Z (2021) Mendeleev Commun 31:498
Ji J, Zhai L, Sun J, He L, Ji J, Ma X, Liu Y, Tang D, Mu Y, Gao Y, Yang H, Iqbal Z, Yang Z (2021) J Heterocycl Chem 58:2390
Fujiu M, Yokoo K, Aoki T, Shibuya S, Sato J, Komano K, Kusano H, Sato S, Ogawa M, Yamawaki K (2020) J Org Chem 85:9650
Garigipati RS (1990) Tetrahedron Lett 31:1969
Gielen H, Alonso-Alija C, Hendrix M, Niewoehner U, Schauss D (2002) Tetrahedron Lett 43:419
Dierks A, Tçnjes J, Schmidtmann M, Christoffers J (2019) Chem Eur J 25:14912
Aurelio L, Box JS, Brownlee RTC, Hughes AB, Sleebs MM (2003) J Org Chem 68:2652
Yin J, Weisel M, Ji Y, Liu Z, Liu J, Wallace DJ, Xu F, Sherry BD, Yasuda N (2018) Org Process Res Dev 22:273
Wikler MA (2009) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, vol 29. Clinical and Laboratory Standards Institute, Wayne
Acknowledgements
Authors thank Ministry of Science and Technology, P.R. China for the award of foreign expert program to Dr. Haikang Yang and Dr. Zafar Iqbal. This work was supported by the grant from Science and Technology Department of Ningxia, P.R. China (No. 2018BCG01001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, Y., Ji, J., Sun, J. et al. Substituted-amidine derivatives of diazabicyclooctane as prospective β-lactamase inhibitors. Monatsh Chem 153, 301–309 (2022). https://doi.org/10.1007/s00706-021-02888-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-021-02888-3