
Abstract
A new ONO-tridentate Schiff base ligand (H2L) derived from 3-methoxysalicylaldehyde and nicotinic hydrazide was synthesized and characterized by elemental analysis, FT-IR, 1H NMR, 13C NMR, UV–Vis, and powder XRD studies. Then, oxovanadium(V) and dioxomolybdenum(VI) Schiff base complexes, VOL and MoO2L, were also prepared and characterized by different techniques. Moreover, the catalytic activities of both complexes were investigated for the synthesis of benzimidazoles, benzoxazoles, and benzothiazoles under reflux conditions as well as through ultrasonic irradiation. The results revealed several advantages of this procedure, including high product yields, short reaction times, facile work-up procedure, simplicity in operation, eco-friendly reaction conditions, and green aspects by avoiding toxic catalysts and solvents.
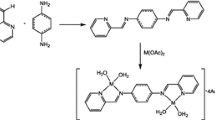
Graphic abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The heterocyclic species consisting of nitrogen atoms constitute the most widespread category of natural compounds and played a very important role in metabolic processes taking place in living organisms [1]. The most salient nitrogen-containing aromatic compounds are benzimidazoles (BIZs), benzoxazoles (BOZs), and benzothiazoles (BTZs) which are available in many natural products. These heteroaromatics are especially found in the molecules which are responsible to carry out the life cycles in various cells and the best example for this is the presence of methyl-substituted benzimidazole motifs in vitamin B-12 [2, 3]. The BIZs, BOZs, and BTZs are simple aromatic compounds consisting of a benzene ring fused with a five-membered imidazole/oxazole/thiazole ring containing heteroatoms at specified positions.
Thiabendazole, a famous fungicidal and antiparasitic agent, is declared as the first available BIZ-based drug [4]. Many derivatives of BIZs, BOZs, and BTZs were developed which showed a broad spectrum of pharmacological activities like antihistamine, anti-ulcerative, antihypertensive, antitumor, antifungal, antiviral, analgesic, anticancer, HIV, antihistaminic, and antiemetic effects [3, 5,6,7,8,9].
Although BIZs, BOZs, and BTZs are very important from a medicinal point of view, still there are a very limited number of methods available in the literature to develop their derivatives. The most conventional synthesis employs the condensation between 1,2-diaminobenzene or 2-aminophenol or 2-aminothiophenol, and carboxylic acids/aldehydes/alcohols or their derivatives under drastic conditions [10,11,12,13,14,15]. A dehydrogenating regime is required which is usually provided with the help of catalysts that are mostly strong acids such as polyphosphoric acid, boric acid, and some mineral acids [16,17,18]. An alternative route is a two-step reaction in which the cyclodehydration of a mono-acylated motif is conducted under various conditions [6].
In addition, some new methods were introduced to prepare derivatives of BIZs, BOZs, and BTZs which include the use of erbium(III) trifluoromethanesulfonate [19], Co(II) salts [14, 20], magnetic nanoparticles with ionic liquids [21], ZnO nanoparticles [22, 23], nanoporous aluminosilicates [24], ionic liquids supported over graphene sheets [15, 25], metallovesicles [26], mesoporous organosilica [27], and mesoporous TiO2-Al2O3 [28]. Anyhow, most of them so far reported synthetic routes have plenty of limitations like (a) rigorous reaction conditions, ((b) employing corrosive organic solvents, (c) multistep procedures, (d) monotonous root arch, (e) poor atom economy, and (f) high temperature. Moreover, the principal pitfall of numerous existing protocols is the catalyst deactivation at the end of synthesis [6, 18]. Many of the endeavors have been carried out to work for the development of feasible, cheaper, cleaner, and one-step methods.
Ultrasonic waves are very well known to play a vital role in the selective synthesis of organic compounds. The spectacular effects of the ultrasonic waves are due to the cavitation phenomena which is associated with some extreme affects, such as local increase of temperature, and local high pressure which leads to the production of free radicals [29] and intense liquid microflows [30]. Ultrasound can be successfully utilized to enhance liquid–solid mass transfer and cause considerable physicochemical changes in the medium [31], which enhances the selectivity as well as the conversion of the desired product by enhancing the rate of the reaction [32]. Hence, ultrasonic irradiations are usually employed for catalytic oxidation reactions.
The chemistry of vanadium and molybdenum has got special attention due to their recent significance in various catalytic processes [33,34,35]. The important chemical reactions catalyzed by oxovanadium and dioxomolybdenum complexes include halo peroxidation, phosphorylation, epoxidation, sulfoxidation, oxygen atom transfer reactions and neutral esterification as well [36,37,38,39,40,41]. In this context, oxovanadium and dioxomolybdenum complexes with hydrazones emerged as fascinating catalysts for numerous liquid phase organic transformations, due to the strong Lewis acid nature under various conditions [1, 41,42,43,44].
Hence, in continuation to our efforts to investigate for new and effective catalysts [45,46,47,48,49], and to look for the catalytic potential of VO(V) and MoO2(VI) complexes for the syntheses of BIZs, BOZs and BTZs, and their derivatives, in this paper, we intend to report the two new VO(V) and MoO2(VI) complexes with hydrazones. The key point of the present work is to validate an effortless and environmentally sound ultrasonic procedure for catalytic synthesis.
Results and discussion
A tridentate Schiff base ligand (H2L) was prepared via the reaction of equimolar amounts of nicotinic hydrazide with 3-methoxysalicylaldehyde in alcohol. Reaction of VO(acac)2 and MoO2(acac)2 with H2L in methanol produces the targeted metal complexes (Scheme 1).

FT-IR study
The stacked FT-IR spectra of the synthesized compounds are shown in Fig. 1. Careful inspection of the spectra is usually accomplished to investigate the site/s of coordination of ligand with metal centers. The two prominent bands in the spectrum of H2L, in the region of 3545 and 1656 cm−1, assigned to the stretching vibrations of the ν(NH) and ν(C = O), disappeared from this specific region upon complexation with metals. These particular sites of attachment are in accordance with the enolization of the amide functional group. This enolization is further confirmed by the emergence of two new bands at 1261 and 1251 cm−1 which are attributed to the enolic ν(C-O) moiety in molybdenum and vanadium complexes, respectively. The azomethine linkage (–HC = N) showed its specific peak at 1604 cm−1 which is also shifted to a higher wavenumber upon attachment with metals. Furthermore, the surfacing of two new bands at 906 and 937 cm−1 are assigned to symmetric and asymmetric stretching vibrations of the cis-Mo(O)2 moiety which are in accordance with similar structures reported previously in the literature [50, 51]. Likewise, the oxovanadium complex also gives its characteristics peak of V = O at 991 cm−1 which is very close to the values for the analogous oxovanadium Schiff base complexes already reported earlier [52,53,54]. Moreover, some new M–O and M–N peaks are also visible at 603 and 457 cm−1 for Mo complex and at 582 and 470 cm−1 for V complex, respectively, which are also in accord with the similar complexes reported formerly [52,53,54,55].

FT-IR spectra of the ligand and its V and Mo complexes
1H and 13C NMR studies
The 1H and 13C NMR spectra of H2L, VOL, and MoO2L recorded in DMSO-d6 are presented in the experimental section and are also shown in Figs. 2, 3, 4, 5, 6 and 7. Two signals appearing at δ = 12.22 and 10.79 ppm in the 1H NMR spectrum of the ligand, corresponding to OH (phenolic) and NH protons, respectively, disappear on treatment with V and Mo salts, demonstrating that the phenolate and enolate oxygens are the sites of coordination of ligand with the metals. This also confirms the occurrence of keto-imine tautomerism upon complexation. Moreover, a singlet signal of azomethine proton (–HC = N) at 8.68 ppm observed in the spectrum of the ligand was shifted downfield at 8.98 ppm showing the deshielding due to the decrease in the electronic density upon the coordination of azomethine nitrogen with the metal center. This is in accordance with the FT-IR spectrum of the complex, where ν(HC = N) appears at a higher wavenumber in comparison with the corresponding free ligand. All the signals of aromatic protons were observed in the expected range of 6.88–9.09 ppm. There is a slight shift in the positions of aromatic protons signals of the ligand upon complex formation (δ = 7.03–9.15 ppm in VOL and 7.05–9.15 ppm in MoO2L).

1H NMR spectrum of the H2L ligand in DMSO-d6

13C NMR spectrum of the H2L ligand in DMSO-d6

1H NMR spectrum of the VOL complex in DMSO-d6

13C NMR spectra of the VOL complex in DMSO-d6

1H NMR spectra of the MoO2L complex in DMSO-d6

13C NMR spectra of the MoO2L complex in DMSO-d6
The 13C NMR spectra of the ligand and its V and Mo complexes are shown in Figs. 3, 5, and 7. The signals for the carbonyl, phenolic, and methine carbon in the V complex were observed at δ = 167.2, 147.7, and 156.9 ppm, respectively, while in the Mo complex these were observed at 167.2, 148.4, and 156.8 ppm, respectively. The chemical shifts of the carbons present in the vicinity of the coordinating atoms (i.e., C8, C1, and C7) showed appreciable changes in their positions due to coordination-induced shifts confirming the participation of these functionalities in coordination. The other aromatic carbons of the ligand and its complexes appeared in their respective regions according to the literature.
UV–Vis study
The electronic transitions for the Schiff base ligand and its complexes were recorded in dimethylformamide (DMF) medium from 270 to 500 nm at room temperature. The electronic spectra are displayed in Fig. 8. The Schiff base, H2L, displayed electronic transitions at 285, 300, 330, and 400 nm. These electronic bands may be attributed to π → π* and n → π* electronic transitions. Upon coordination to vanadium and molybdenum metals, a blue shift is observed in the UV–Vis spectra of the complexes and the UV pattern of both complexes cross each other at approximately 300 nm. The emergence of the new optical band around 400 nm may be assigned as phenoxo to metal ion electronic transition [56,57,58,59].

UV–Vis spectra of the Schiff base ligand and its complexes in DMF medium
Powder XRD study
The phase analysis and crystalline structure of the ligand and two complexes were further confirmed through powder X-ray diffraction (PXRD). A scan from a low angle and short-range with 2θ = 5–20 degree resulted in distinct PXRD spectra of these three compounds. The main difference lies in terms of the reduced relative intensities for complexes in comparison to that for ligand. This could be attributed to the loss of coordinated solvent molecules during complex formation and the coordination of bulky metal atoms. Further, the PXRD diffractions observed at around 2θ = 7 and 9 degree with large intensities for both complexes seems a characteristic for Mo and V bonded with oxygen atoms [60] Fig. 9.

PXRD patterns of the Schiff base ligand and its complexes
Catalytic activity studies
After successful syntheses and characterization of VOL and MoO2L, their catalytic activities were evaluated for the synthesis of benzimidazoles, benzoxazoles, and benzothiazoles. To optimize the reaction conditions, the reaction between 1,2-phenylenediamine and benzaldehyde (1:1 molar ratio) for the synthesis of 2-phenylbenzimidazole was selected as a model reaction, and the effect of solvent and the amount of catalyst were investigated. As shown in Table 1, water and various organic solvents such as ethanol, methanol, acetonitrile, DMSO, 1,2-dichloroethane, chloroform, and dichloromethane were examined in the presence of 10 mol% of the catalyst and it was found that ethanol was the best solvent for the reaction. Next, the effect of the catalytic amount on the time and yield of the reaction was investigated using different amounts of VOL and MoO2L. The best result was obtained with 10 mol% of the catalyst. It is noteworthy that in the absence of a catalyst, no product was formed even after the period of 24 h (Table 1, entry 1).

Due to the great popularity of ultrasound in chemical syntheses, which reduces the reaction time and increases the yield of products, and for eco-friendly reasons, this reaction was also conducted under ultrasonic irradiation. The results showed that the reaction was completed in a short time along with a higher isolated yield in the presence of both, VOL and MoO2L complexes (Table 1, entry 15).
It is noteworthy that in all cases, only 2-phenylbenzimidazole was produced. The disubstituted benzimidazole (1-benzyl-2-phenylbenzimidazole) was obtained only if two equivalents of benzaldehyde were employed in aqueous media under reflux conditions.
To explore the generality and versatility of this procedure, different substituted aryl aldehydes were condensed with 1,2-phenylenediamine for the synthesis of corresponding benzimidazole derivatives. Aryl aldehydes containing both electron-donating and electron-withdrawing groups were used and as expected, they gave well to the excellent yield of products (Table 2, entries 1–13). Under similar conditions, the methodology was extended to the synthesis of various benzoxazoles and benzothiazoles from 2-aminophenol and 2-aminothiophenol, respectively (Table 2, entries 14–29).

Table 3 compares the catalytic efficiencies of VOL and MoO2L with some reported catalysts in the condensation of 1,2-phenylenediamine with benzaldehyde to produce 2-phenyl-1H-benzoimidazole (5a). It can be concluded from Table 3 that many of the previously reported procedures are associated with numerous drawbacks such as necessity of surplus quantity of catalyst, elevated temperature, reduced yield, lengthy reaction time, and employ the use of volatile and noxious organic solvents. Thus, the present technique overcomes the drawbacks of the previously reported methodologies and has dominance due to high yield, short reaction time, and facile synthetic conditions.
The proposed mechanistic pathway for the preparation of benzimidazoles in the presence of VOL or MoO2L catalyst is presented in Scheme 2. Presumably, the Schiff base complex acting as a Lewis acid catalyst, is coordinated with the carbonyl group of aldehyde and then the activated carbonyl group is attacked by 1,2-phenylenediamine to obtain intermediate I. This intermediate loses water in an elimination reaction in the presence of a catalyst to imine II. Intramolecular nucleophilic addition and then proton transfer causes ring closure and gives compound III, which is oxidized upon exposure to air and gives the corresponding benzimidazole. A similar mechanism can be drawn for the synthesis of benzoxazoles and benzothiazoles catalyzed by VOL and MoO2L complexes.

The recyclability and reusability of the V and Mo complexes were studied for the reaction of benzaldehyde and 1,2-phenylenediamine under ultrasonic irradiation at room temperature. On the completion of reaction, the reaction mixture was filtered and the precipitates were washed with ethanol to obtain the pure products. The catalyst was recycled by evaporating the ethanolic phase in air and washing again with cold methanol. After being air dried, the recycled catalyst could be reused as such in subsequent experiments under similar reaction conditions. The yields of the product remained comparable in all experiments, indicating that the catalyst can be recycled at least five times with no considerable loss in its activity (Table 4).
Conclusion
In this research work we have synthesized a new tridentate ONO-donor Schiff base ligand and its oxovanadium and dioxomolybdenum complexes, VOL and MoO2L, and characterized them by elemental analysis, FT-IR, 1H NMR, 13C NMR, UV–Vis, and powder XRD studies. Moreover, the catalytic activity studies of the complexes have also been performed in the synthesis of benzimidazoles, benzoxazoles, and benzothiazoles under conventional heating and ultrasonic irradiation. The short reaction span, improved yield, and mild reaction conditions are the main edges of the ultrasonic methodology.
Experimental
All chemicals were of reagent grade and used without further purification. Elemental analyses were performed with a Heraeus CHN-O-FLASH EA 1112 instrument. 1H and 13C NMR spectra were recorded at ambient temperature with a BRUKER AVANCE 400 MHz spectrometer using tetramethylsilane (TMS) as an internal standard reference. The chemical shift values (δ) are given in ppm. Infrared spectra were recorded using KBr pellets on a IR Prestige-21 spectrophotometer. The electronic absorption spectra of the ligand and its complexes in DMF were recorded by a UV- 1601 PC spectrophotometer. The XRD patterns were recorded using a Bruker D8 ADVANCE X–ray diffractometer equipped with nickel monochromatized CuKα radiation (λ = 1.5406 Å). A UP 400S ultrasonic processor equipped with a 3 mm wide and 140 mm long probe, which was immersed directly into the reaction mixture, was used for sonication. The reactions under ultrasonic irradiation were carried out at room temperature in a 40 cm3 glass reactor.
(E)-N′(2-Hydroxy-3-methoxybenzylidene)nicotinohydrazide (H2L, C14H13N3O3)
Nicotinic hydrazide (1.37 g, 10 mmol), dissolved in 25 cm3 of hot methanol, was added dropwise to a solution of 1.52 g 3-methoxysalicylaldehyde (10 mmol) in 25 cm3 of methanol. The reaction mixture was refluxed for about 3 h using a magnetically equipped hot plate. Upon cooling to room temperature, the resulting precipitates were collected by suction filtration and washed thrice with cold methanol to get the desired product H2L. The Schiff base ligand was spectroscopically pure and used in the syntheses of the corresponding metal complexes. White solid; yield 87%; FT-IR (KBr): \(\bar{\nu }\)= 3545 (\(\nu\) N–H), 1656 (\(\nu\) C=O), 1604 (\(\nu\) C=N), 1571, 1475 (\(\nu\) C=C), 1247 (\(\nu\) C-O), 1076 (\(\nu\) N–N) cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 3.83 (3H, s, H-C14), 6.88 (1H, H-C4, t, 3 J = 7.8 Hz), 7.05 (1H, H-C3, d, 3 J = 7.8 Hz), 7.19 (1H, H-C5, d, 3 J = 7.8 Hz), 7.59 (1H, H-C12, dd, 3 J = 7.8 Hz, 3 J = 4.8 Hz), 8.31 (1H, H-C13, dt (br), 3 J = 7.8 Hz), 8.68 (1H, s, CH = N), 8.78 (1H, H-C11, dd, 3 J = 4.7 Hz, 4 J = 1.3 Hz), 9.09 (1H, H-C10, d, 4 J = 1.6 Hz), 10.79 (1H, NH, s), 12.22 (1H, OH, s) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 55.8 (C14), 113.9 (C4), 118.9 (C3), 119.1 (C6), 120.5 (C5), 123.6 (C12), 128.7 (C9), 135.4 (C13), 147.1 (C1), 147.9 (C2), 148.4 (C7), 148.6 (C10), 152.4 (C11), 161.4 (C8) ppm.
(N-[(3-Methoxy-2-oxidophenyl)methylidene]pyridine-3-carbohydrazonato)-bis(oxido)-methoxo-oxo-vanadium(V) (VOL, C15H14N3O5V)
The new VOL complex was prepared by mixing 0.265 g of [VIVO(acac)2] (1 mmol, acac = acetylacetonate) with 0.271 g of H2L (1 mmol) in 50 cm3 of methanol and then keeping the reaction mixture under reflux conditions for 3 h. The solid product obtained was filtered off, washed carefully with water, methanol, and diethyl ether and finally dried in vacuo. Green solid; yield 65%; FT-IR (KBr): \(\bar{\nu }\)= 1604 (\(\nu\) C=N), 1352 (\(\nu\) C=N–N=C), 1473, 1550 (\(\nu\) C=C), 1251 (\(\nu\) C-O), 1039 (\(\nu\) N–N), 991 (\(\nu\) V=O), 582 (\(\nu\) V–O), 470 (\(\nu\) V–N) cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 3.17 (3H, -OCH3 coord., s), 3.83 (3H, s, H-C14), 7.03 (1H, H-C4, t, 3 J = 7.9 Hz), 7.26 (1H, H-C3, d, 3 J = 7.9 Hz), 7.34 (1H, H-C5, d, 3 J = 7.9 Hz), 7.57 (1H, H-C12, dd, 3 J = 7.9 Hz, 3 J = 4.8 Hz), 8.32 (1H, H-C13, dt, 3 J = 7.9 Hz, 4 J = 1.7 Hz), 8.76 (1H, H-C11, d, 3 J = 4.8 Hz), 8.98 (1H, CH = N, s), 9.15 (1H, H-C10, s) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 55.8 (C14), 64.6 (C, MeOH coord.), 119.0 (C4), 120.7 (C6), 121.7 (C3), 123.9 (C5), 125.7 (C12), 126.2 (C9), 135.5 (C13), 147.7 (C1), 148.8 (C10), 149.5 (C2), 152.5 (C11), 156.9 (C7), 167.2 (C8) ppm.
(N-[(3-Methoxy-2-oxidophenyl)methylidene]pyridine-3-carbohydrazonato)-bis(oxido)-methanol-molybdenum(VI) (MoO2L, C15H15MoN3O6)
In a round bottom flask equipped with a magnetic stirrer bar, 0.271 g of the Schiff base H2L (1 mmol) and 0.330 g of [MoVIO2(acac)2] (1 mmol) were suspended in 50 cm3 of methanol. The resulting suspension was stirred under reflux conditions for 3 h. After that, two-thirds of the solvent was evaporated and the solution was cooled over an ice bath to decrease the solubility of dissolved contents. The resultant solid product was filtered off and washed multiple times with water, methanol, diethyl ether, and eventually dried in vacuo. Orange solid; yield 78%; FT-IR (KBr): v̄ = 1620 (\(\nu\) C=N), 1435 (\(\nu\) C=N–N=C), 1469, 1564 (\(\nu\) C=C), 1261 (\(\nu\) C-O), 1047 (\(\nu\) N–N), 937 (\(\nu\) O=Mo=O) asym, 906 (\(\nu\) O=Mo=O) sym, 603 (\(\nu\) Mo–O), 457 (\(\nu\) Mo–N) cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 3.18 (3H, -CH3 MeOH coord., d, 3 J = 6.9 Hz), 3.82 (3H, s, H-C14), 4.08 (1H, -OH MeOH coord., q, 3 J = 6.9 Hz), 7.05 (1H, H-C4, t, 3 J = 7.9 Hz), 7.26 (1H, H-C3, dd, 3 J = 7.9 Hz, 4 J = 1.0 Hz), 7.33 (1H, H-C5, dd, 3 J = 7.9 Hz, dd, 4 J = 1.0 Hz), 7.56 (1H, H-C12, dd, 3 J = 7.8 Hz, 3 J = 4.8 Hz), 8.31 (1H, H-C13, dt, 3 J = 8.0 Hz, 4 J = 1.8 Hz), 8.76 (1H, H-C11, d, 3 J = 3.7 Hz), 8.98 (1H, CH = N, s), 9.15 (1H, H-C10, br) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 55.8 (C14), 65.6 (C, MeOH coord.), 117.3 (C4), 120.4 (C6), 121.6 (C3), 124.0 (C5), 125.4 (C12), 126.1 (C9), 135.4 (C13), 148.4 (C1), 148.7 (C10), 149.2 (C2), 152.4 (C11), 156.8 (C7), 167.2 (C8) ppm.
General experimental procedure under conventional heating method
To a solution of 1,2-phenylenediamine, 2-aminophenol, or 2-aminothiophenol (1 mmol) and aryl aldehydes (1 mmol) in 5 cm3 of EtOH, VOL or MoO2L complexes (10 mol%) was added and the mixture was stirred under reflux conditions for the specified intervals of times. The progress of the reaction was continuously monitored by TLC (eluent n-hexane: ethyl acetate, 5:2). On completion of the reaction, the mixture was cooled down over ice, filtered off and recrystallized from the ethanol to afford the pure respective products. All of the obtained products are known and identified by comparing their physicochemical and spectroscopic data (FT-IR, NMR) with those of authentic samples.
General experimental procedure under ultrasound-assisted method
10 mol% of the catalyst, VOL or MoO2L, was added to an ethanolic solution of aryl aldehydes (1 mmol) and 1,2-phenylenediamine, 2-aminophenol, or 2-aminothiophenol (1 mmol). The mixture was subjected to ultrasonic irradiation at room temperature for an appropriate period of time until the reaction was completed, as indicated by TLC (eluent n-hexane: ethyl acetate, 5:2). Then, the reaction mixture was filtered and the product was finally recrystallized from the ethanol to achieve maximum purity.
References
Kebede E, Tadikonda R, Nakka M, Inkollu B, Vidavalur S (2015) Eur J Org Chem https://doi.org/10.1002/ejoc.201500803
Alaqeel SI (2017) J Saudi Chem Soc 21:229
Di Gioia ML, Cassano R, Costanzo P, Herrera Cano N, Maiuolo L, Nardi M, Nicoletta FP, Oliverio M, Procopio A (2019) Molecules 24:2885
Yadav G, Ganguly S (2015) Eur J Med Chem 97:419
Algul O, Karabulut A, Canacankatan N, Gorur A, Sucu N, Vezir O (2012) Med Chem 11:267
Sharghi H, Asemani O, Tabaei SMH (2008) J Heterocycl Chem 45:1293
Stuchlikova L, Jirasko R, Skalova L, Pavlik F, Szotakova B, Holcapek M, Vanek T, Podlipna R (2016) Chemosphere 157:10
Ranjan N, Kumar S, Watkins D, Wang D, Appela DH, Arya DP (2013) Bioorg Med Chem Lett 23:5689
Roth T, Morningstar ML, Boyer PL, Hughes SH, Buckheit RW, Michejda CJ (1997) J Med Chem 40:4199
Chi Y-C, Sun CM (2000) Synlett 5:591–594
Dudd LM, Venardou E, Garcia-Verdugo E, Licence P, Blake AJ, Wilson C, Poliakoff M (2003) Green Chem 5:187
Bahrami K, Khodaei MM, Kavianima I (2007) Synthesis 417
Sharghi N, Asemani O, Khalifeh R (2008) Synth Commun 38:1128
Narang U, Yadav KK, Bhattacharya S, Chauhan SM (2017) ChemistrySelect 2:7135
Nguyen TT, Nguyen XT, Nguyen TL (2019) ACS Omega 4:368
Perston PN (1981) The Chemistry of Heterocyclic Compounds, Benzimidazoles and Congeneric Tricyclic Compounds, Part 1. Wiley, New York, 40:6
Grimmett MR (1984) Imidazoles and their benzo derivatives. In: Katritzky AR, Rees CW (eds) Comprehensive Heterocyclic Chemistry, vol 5. Pergamon Press, Oxford, p 457
Gadekar SL, Arbad RB, Machhindra KL (2010) Chin Chem Lett 21:1053
Cano NH, Uranga JG, Nardi M, Procopio A, Wunderlin DA, Santiago AN (2016) Beilstein J Org Chem 12:2410
Khan AT, Parvin T, Choudhury LH (2009) Synth Commun 39:2339
Rezaee Nezhad E, Tahmasebi R (2019) Asian J Green Chem 3:34
Alinezhad H, Salehian F, Biparva P (2012) Synth Commun 42:102
Nale DB, Bhanage B (2015) Synlett 2835
Chari MA, Shobha D, Kenawy E, Al-Deyab SS, Reddy BVS, Vinu A (2010) Tetrahedron Lett 51:5195
Hanoon HD, Kowsari E, Abdouss M, Zandi H, Ghasemi MH (2017) Res Chem Intermed 43:1751
Kaur N, Kaur S, Kaur G, Bhalla A, Srinivasan S, Chaudhary GR (2019) J Mater Chem A 7:17306
Haghighat M, Golshekan M, Shirini F (2019) ChemistrySelect 4:7968
Bahrami K, Bakhtiarian M (2018) ChemistrySelect 3:10875
Kelkar MA, Gogate RP, Pandit AB (2006) Ultrason Sonochem 13:523
Margulis MA (1995) Sonochemistry and Cavitation. Australia ; [Langhorne, Pa., U.S.] : Gordon and Breach Publishers
Moghadam M, Mirkhani V, Tangestaninejad S, Mohammadpoor-Baltork I, Kargar H (2009) J Iran Chem Soc 6:251
Mahamuni NN, Gogate RP, Pandit AB (2007) Ultrason Sonochem 14:135
Heidari F, Fatemi SJA, Ebrahimipour SY, Ebrahimnejad H, Castro J, Dusek M, Eigner V (2017) Inorg Chem Commun 76:1
Quintal S, PiresdaSilva MJ, Martins SRM, Sales R, Felix V, Drew MGB, Meirels M, Mourato AC, Nunes CD, Saraiva MS, Machuqueiro M, Calhorda MJ (2019) Dalton Trans 48:8449
Pisk J, Bilić L, Đaković M, Cvijanović D, Damjanović V, Lovrić J, Rubčić M, Vrdoljak V, Cindrić M (2018) Polyhedron 145:70
Nag P, Bohra R, Mehrotra RC (2002) J Chem Res 86
Zhang WG, Liang JH (2017) Russ J Coord Chem 43:411
Hossain MK, Haukka M, Sillanpää R, Hrovat DA, Richmond MG, Nordlander E, Lehtonen A (2017) Dalton Trans 46:7051
Yang Y, Hao S, Zhang Y, Kan Q (2011) Solid State Sci 13:1938
Ta S, Ghosh M, Ghosh K, Brandão P, Félix V, Hira SK, Manna PP, Das D (2019) ACS Appl Bio Mater 2:2802
Wang Q, Xiong ZD, Liu L, Cai YJ (2021) Inorg Nano-Met Chem 51:12
Safaiee M, Moeinimehr M, Zolfigol MA (2019) Polyhedron 170:138
Liang M, Sun N, Zou D-H (2018) Acta Chim Slov 65:964
Xue L-W, Peng Q-L, Wang P-P, Zhang H-J (2019) Acta Chim Slov 66:694
Hatefi M, Moghadam M, Sheikhshoaei I, Mirkhani V, Tangestaninejad S, Mohammadpoor-Baltork I, Kargar H (2009) Appl Catal A Gen 370:66
Moghadam M, Mirkhani V, Tangestaninejad S, Mohammadpoor-Baltork I, Kargar H, Sheikhshoaei I, Hatefi M (2011) J Iran Chem Soc 8:1019
Kargar H (2011) Inorg Chem Commun 14:863
Moshref Javadi M, Moghadam M, Mohammadpoor-Baltork I, Tangestaninejad S, Mirkhani V, Kargar H, Tahir MN (2014) Polyhedron 72:19
Kargar H (2014) Transit Met Chem 39:811
Vrdoljak V, Prugovecki B, Calogovic DM, Pisk J, Dreos R, Siega P (2011) Cryst Growth Des 11:1244
Kia R, Kargar H (2015) J Coord Chem 68:1441
Kurbah SD, Asthana M, Syiemlieh I, Lywait AA, Longchar M, Lal RA (2018) J Organomet Chem 876:10
Li Y, Xu L, Duan M, Wu J, Wang Y, Dong K, Han M, You Z (2019) Inorg Chem Commun 105:212
Gao S, Zhang XF, Huo LH, Zhao H (2004) Acta Crystallogr E60:m1731
Rana A, Dinda R, Sengupta P, Ghosh S, Falvello LR (2002) Polyhedron 21:1023
Mahato S, Meheta N, Kotakonda M, Joshi M, Ghosh P, Shit M, Choudhury AR, Biswas B (2020) Appl Organomet Chem 34:e5935
Mahato S, Meheta N, Kotakonda M, Joshi M, Shit M, Choudhury AR, Biswas B (2020) Polyhedron 194:114933
Mudi PK, Bandopadhyay N, Joshi M, Shit M, Paul S, Choudhury AR, Biswas B (2020) Inorg Chim Acta 505:119468
Pal CK, Mahato S, Joshi M, Paul S, Choudhury AR, Biswas B (2020) Inorg Chim Acta 506:119541
Sen SK, Al Mortuza A, Manir MS, Pervez MF, Hossain SMAI, Alam MS, Haque MAS, Matin MA, Hakim MA, Huda A (2020) Nano Ex 1:020026
Fallah-Mehrjardi M, Ayazi M, Banitaba SH (2020) Iran Chem Commun 8:80
Teimouri A, Chermahini AN, Salavati H, Ghorbanian L (2013) J Mol Catal A Chem 373:38
Kumar KR, Satyanarayana PVV, Reddy BS (2013) J Chem 2013:151273
Digwal CS, Yadav U, Sakla AP, Ramya PVS, Aaghaz S, Kamal A (2016) Tetrahedron Lett 57:4012
Xiangming H, Huiqiang M, Yulu W (2007) ARKIVOC xiii:150
Chari MA, Shobha D, Sasaki T (2011) Tetrahedron Lett 52:5575
Sadeghi B, Ghasemi Nejad M (2013) J Chem 2013:581465
Inamdar SM, More VK, Mandal SK (2013) Tetrahedron Lett 54:579
Azizian J, Torabi P, Noei J (2016) Tetrahedron Lett 57:185
Bharathi M, Indira S, Vinoth G, Mahalakshmi T, Induja E, Bharathi KS (2020) J Coord Chem 73:653
Acknowledgements
We gratefully acknowledge the practical support of this study by Ardakan University and Payame Noor University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kargar, H., Kargar, K., Fallah-Mehrjardi, M. et al. Syntheses, characterization, and catalytic potential of novel vanadium and molybdenum Schiff base complexes for the preparation of benzimidazoles, benzoxazoles, and benzothiazoles under thermal and ultrasonic conditions. Monatsh Chem 152, 593–605 (2021). https://doi.org/10.1007/s00706-021-02780-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-021-02780-0