
Abstract
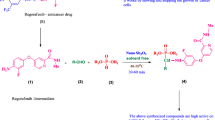
A straight forward approach ensuring the anchoring of bisphosphonate moiety onto various pyridinyl and pyrimidinyl amines is described by the three-component reaction of an amine, diethyl phosphite, and triethyl orthoformate in the presence of catalytic amount of nano ZnO as an environmentally benign and heterogeneous catalyst under solvent-free microwave irradiation conditions. All the synthesized compounds are evaluated for anti-cell proliferation activity against human breast (MCF-7), prostate (DU-145), osteosarcoma (MG-63), fibrosarcoma (HT-1080), multiple myeloma (RPMI-8226) cancer cell lines using sulforhodamine-B (SRB) assay method, and adriamycin as a reference drug. All the title compounds showed promising cytotoxic activity on the five cell lines. One compound was two times more active than adriamycin on all the five cancer cell lines.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heteroaromatic compounds those bearing nitrogen as a heteroatom draw an enormous interest due to their wide-range spectrum of biological and pharmaceutical applications [1,2,3,4]. The pyridine and pyrimidine moieties are common core heterocyclic motifs present in some well-known components of human organisms [5,6,7,8]. Pyridine derivatives are an important class of aza-heterocycle occurred in many natural products and active pharmaceuticals, including vitamin B3 (niacin), vitamin B6 (pyridoxine), nicotine, isoniazid, and other nitrogen-containing plant products [9]. Pyrimidines are very important biologically active heterocycles that represent the most abundant fellows of the diazine family with cytosine, uracil, and thymine being constituents of ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). In addition to this, pyrimidine moieties also occur in many natural products such as vitamin B1 (thiamine) and many pharmaceutical compounds, such as barbituric acid and the HIV drug zidovudine [10].
Aminomethylene bisphosphonates are well established in the management of cancer-induced skeletal disorders [11]. Recent research studies suggest that nitrogen-containing bisphosphonates (N-BPs) stimulate the apoptosis of tumor cells as well as osteoclasts in bone metastatic sites [12]. They are potent inhibitors of osteoclast-mediated bone resorption and play a vital role in the supportive care of patients with bone metastases [13]. Several N-BPs such as clodronate, pamidronate, and zoledronic acid develop the clinical outcome for patients with multiple myeloma [14] and metastatic breast cancer [15]. Recently, zoledronic acid was found to increase disease-free survival and overall survival in some adjuvant breast cancer settings and prolonged survival in patients with several advanced cancers [16]. Besides the bone resorption and antitumor activity of bisphosphonates, they are also showing antioxidant [17], antimicrobial [18], antitrypanosomal [19, 20], and herbicidal [21] activities. Along with their biological importance, aminomethylene bisphosphonates are also known for their metal-chelating ability. The uranyl (UO2 2+)-binding properties of these compounds were listed [22] to have excellent association constants.
The common synthetic routes to aminomethylene bisphosphonates (N-BPs) involve acid-catalyzed reactions of nitriles with phosphorous acid or phosphites, Beckmann rearrangement of oximes in the presence of phosphites [23], and reductive amination of carbonyl derivatives with aminomethyl diphosphonate [24]. However, the aforementioned procedures have one or more drawbacks such as long reaction times, require stoichiometric quantities of toxic catalysts, give product yields poor, and generate vast amounts of wastage [25].
It is evident from the recent literature that nanochemistry is an up growing research area due to their unique properties. These materials exhibit better catalytic activity compared to their bulk-sized counterparts [26,27,28]. The usage of nanomaterial provides advantages of high atom efficiency, simplified isolation of product, easy recovery, and recyclability of the catalysts. Among the nanoparticles, zinc oxide (nano ZnO) has received considerable attention due to its low toxicity, cost effectiveness, air and water compatibility, ease of handling, good reactivity, recyclability, experimental simplicity, remarkable ability to suppress side reactions in acid-sensitive substrates, environment friendly, and many other potential applications in diverse fields [29,30,31,32,33,34,35].
In recent days, microwave-assisted organic chemistry has growing at a rapid rate, because this technique provides an effective and alternative energy source for carrying out the chemical reactions and processes [36]. Thus, application of microwave energy to promote organic reactions is of increasing attention and offers numerous advantages over the conventional techniques [37]. Microwave reactions are eco-friendly and find valuable applications in organic synthesis [37,38,39,40], peptide synthesis [41], nanotechnology [42], polymer chemistry [43], and biochemical processes [44, 45]. In this study, we accomplished the synthesis and in vitro anticancer activity studies of tetraethyl (pyridinyl/pyrimidinyl amino)methylene bisphosphonates (TPAM-BPs) using nano ZnO as environmentally benign, reusable, heterogeneous acid catalyst under solvent-free and microwave irradiation conditions.
Results and discussion
A series of aminomethylene bisphosphonates 4a–4j were synthesized via tri-component one-pot neat reaction of various pyridinyl and pyrimidinyl amines (1a–1j), diethyl phosphite (2), and triethyl orthoformate (3) using nano ZnO as an efficient catalyst under solvent-free and microwave irradiation conditions (Scheme 1). The products were obtained in high yields after simple work-up procedure.

In order to determine the best experimental conditions, we accomplished a model reaction by taking 3-aminopyridine (1a; 1 mmol), diethyl phosphite (2; 2.2 mmol), and triethyl orthoformate (3; 1 mmol). Initially, we have run the reaction under solvent-free conditions by using different catalysts such as FeCl3, ZnCl2, I2, CuCl2, AlCl3, p-TSA, BF3·SiO2, and Amberlyst 15 along with nano ZnO at 100 °C, and the results are listed in Table 1. Among all these catalysts, it was found that nano ZnO showed better catalytic activity. Most excitingly, when nano ZnO was used in 7.5 mol%, the reaction proceeded very smoothly and gave the product 4a in 93% yield (Table 1, entry 10).

Further, to determine the exact requirement of catalyst for the reaction, we investigated the model reactions at different concentrations of catalyst such as 1, 2.5, 5, 7.5, and 10 mol% were loaded and yields were 35, 60, 75, 93, and 92%, respectively (Fig. 1). This indicates that 7.5 mol% of nano ZnO is sufficient for the best result by considering the reaction time and yield of product. Further, there is no development in the product yield even though the amount of catalyst is amplified. Thus, we generalized the procedure that 7.5 mol% of nano ZnO is sufficient for the synthesis of aminomethylene bisphosphonates under neat conditions. The catalytic activity of the recycled nano ZnO was also examined. Nano ZnO could be reused five times for the reaction without noticeable loss of activity (Fig. 2). When the effect of microwave irradiation power was studied on this reaction, use of high watts microwave irradiation not only enhanced the reaction rate but also avoided the side reactions.

Optimization of catalyst concentration

Reusability of the catalyst
We investigated the scope and limitations of the catalyst for the reaction using various substituted pyridinyl/pyrimidinyl amines 1a–1j, diethyl phosphite (2), and triethyl orthoformate (3), as shown in Table 2. The experimental results indicated that this catalyst was efficient for all the substituted pyridinyl/pyrimidinyl amines. From the results, we found that the 7.5 mol% of the catalyst nano ZnO, MWI at 100 °C for 5 min without solvent are the optimum conditions for the formation of title compounds 4a–4j in good to excellent yields. High yields of the product, less reaction time, low catalyst loading, and reusability of the catalyst (up to five cycles), and microwave irradiation conditions are the advantages of this method.
Reusability of the catalyst
The recyclability of the nano ZnO catalyst was checked for the model reaction (4a). After each run, dichloromethane (2 × 15 cm3) was added to the reaction mixture and filtered. The catalyst residue was washed with chloroform, dried, and reused for next run. The yields obtained are in 93, 91, 90, 88, and 86% yields over five cycles (Fig. 2). The results indicate that the catalyst can be reused for five times without loss of its activity.
Formation of the title compounds 4a–4j in the presence of nano ZnO (Scheme 2) involves the mechanism of the three-component condensation [47, 48], which is similar to the Kabachnik–Fields reaction in which a carbonyl component is replaced by triethyl orthoformate of corresponding electrophilic character. Imines are commonly suggested as intermediates of the Kabachnik–Fields reaction. The first step of the condensation is the interaction of orthoformate with the nano ZnO surface that generated the more electrophilic carbon center followed by the nucleophilic attack of amine to give imine-type intermediate I. The more electrophilic carbon center generated by catalytic surface was attacked by the nucleophilic amine. In this step, nano ZnO as strong Lewis acid enhances the electrophilicity of the active carbon of the orthoformate. In the next steps, the nucleophilic addition of two moles diethyl phosphite to the C=N bond of the imines results in phosphonates II and III, respectively. Then, the elimination of an ethanol may lead to the formation of bisphosphonates IV.

The compounds 4a–4j were characterized by physical and spectral (IR, NMR, and mass) data. They showed strong IR absorption bands in the regions of 3296–3214, 1264–1224, 1024–1009, and 760–720 cm−1 for –NH, –P=O, –P–O–C, and P–C aliphatic stretching frequencies, respectively. In 1H NMR spectra, the singlet at δ = 6.86–5.82 ppm confirmed the –NH protons. The chemical shifts in the region of 8.95–6.48 ppm are due to aromatic protons and the multiplet at 5.64–4.85 ppm corresponds to HC–P proton. The multiplet in the region of 4.18–4.04 and multiplet at 1.38–1.14 ppm are due to the O–CH 2–CH3 and O–CH2–CH 3. In 13C NMR spectra, the chemical shifts in the region of 184.5–101.3 ppm are assigned to carbons of aromatic ring; the signals in the region of 64.6–58.4, 52.6–43.4, and 16.4–16.1 ppm confirmed the –O–CH 2–CH3, HC–P, and –O–CH2–CH 3 carbons. The 31P NMR chemical shifts of the title compounds appeared in the range of 16.2–24.4 ppm.
In vitro cytotoxic activity
In order to investigate the effectiveness of the in vitro cell cytotoxic properties of synthesized aminomethylene bisphosphonates, they were subjected to the well-known sulforhodamine-B (SRB) cytotoxic assay [49] against human breast (MCF-7), prostate (DU-145), osteosarcoma (MG-63), fibrosarcoma (HT-1080), and multiple myeloma (RPMI-8226) cancer cell lines. All the data were expressed as IC50 values (the dose which causes 50% inhibition of viable cells) of the tested compounds. The results are illustrated in Table 3. Compounds 4i and 4j have the most active and broad spectrum activities on the five cancer cell lines which are more active than that of standard drug adriamycin. Compound 4j was two times more active than that of adriamycin in all the five cell lines. While compounds 4d, 4e, 4f, 4g, and 4h showed moderate cytotoxic activity, the compounds 4b, 4c, and 4a could not show effective cytotoxic activity on all the five cell lines.
In the previous literature [50], adriamycin was used as a standard drug against various cancer cell lines such as colon (SW620), breast (MCF7), cervix (HeLa), and liver (HEPG2), and the values were 35.8, 43.8, 57.2, and 46.6, respectively. Likewise, in another report [51], for colon (SW480), lung carcinoma cells (A549), hepatic carcinoma cells (HepG2), and cervical cancer cells (HeLa), the obtained standard values are 10.9, 13.5, 11.5, and 12.5, respectively. Comparatively the same drug as standard in another report [52] acted against breast (MDA-MB453, MCF7), lung (NCI-H522, NCI-H23), and kidney (HEK-2933T) cancer cell lines and the values are 23.98, 19.01, 25.92, 26.76, and 70.23, respectively. While our compounds when treated for various cancer lines such as human breast (MCF-7), prostate (DU-145), osteosarcoma (MG-63), fibrosarcoma (HT-1080), multiple myeloma (RPMI-8226), using adriamycin as a standard drug, the obtained values are 12.42, 11.57, 8.79, 10.23, and 9.76, respectively. When our compounds 4a–4j are treated with the aforementioned cancer cell lines, all the compounds showed moderate to good activity against all the five cancer call lines, but compound 4j showed (6.15, 5.48, 4.32, 5.47, and 4.63) twice more activity than that of adriamycin in all the five cell lines.
Structure–activity relationship (SAR)
Structure–activity relationship studies showed that the anticancer activity exhibited by the newly synthesized aminomethylene bisphosphonates depended on two series: one is pyridine series (4a–4e) and another one is pyrimidine series (4f–4j). In general, the pyrimidine series exhibited better antitumor activity than pyridine series. Among the pyrimidine series, 4j and 4i exhibited highest tumor activity when compared to adriamycin. This might be due to the presence of the benzene ring and bromine attached to pyrimidine ring, respectively.
Conclusions
In conclusion, nano ZnO was found to be an efficient catalyst for the synthesis of aminomethylene bisphosphonates 4a–4j in good to excellent yields. The notable advantages of the present synthetic protocol are operational simplicity, eco-friendly, catalyst reusability, simple work-up, and high yields. Among the synthesized compounds, 4i and 4j showed potent cytotoxic activity against various cell lines than that of the standard drug Adriamycin. Interestingly, compound 4j is twice more active than the standard drug. We believe that this method serves a practical alternative to existing methods for the synthesis of aminomethylene bisphosphonates.
Experimental
All the solvents and chemicals were procured from Merck and Sigma-Aldrich and were used without further purification. Microwave irradiation was carried out in microwave oven, catalyst system (CATA-4R). Melting points of the compounds were determined in open capillary tube on Guna melting pointing apparatus and are uncorrected. Infrared spectra were recorded on Bruker ALPHA interferometer instrument. 1H, 13C, and 31P NMR were recorded on Bruker 400 MHz instrument. Mass spectra were recorded on ESI–MS model in positive mode and elemental analysis was carried out in FLASH EA Thermo Finnigan 1112 instrument. The cancer cell lines for anticancer activity were obtained from American Type Culture Collection (ATCC, Rockville, MD) and cultured with RPMI 1640 medium (GIBCO BRL, Grand Island, NY, USA).
General procedure for the synthesis of aminomethylene bisphosphonates 4a–4j
Various pyridinyl/pyrimidinyl amines (1a–1j; 1 mmol), diethyl phosphite (2; 2.2 mmol), triethyl orthoformate (3; 1 mmol), and 7.5 mol% of nano ZnO were taken in a flat-bottomed flask and irradiated with microwave irradiation using Catalyst System (CATA-4R) at 400 W. The progress of the reaction was monitored by TLC (3:2; n-hexane:ethyl acetate) for every 1 min. The reaction was completed in 5 min. After completion of reaction, the mixture was dissolved in 10 cm3 of DCM and filtered to remove the catalyst as residue. The organic layer was washed with water (2 × 5 cm3) and the water layer was discarded. The combined organic mixture was washed with 5 cm3 brine solution, dried over anhydrous Na2SO4, and concentrated under reduced pressure; the solid obtained was washed with cold water, air-dried, and recrystallized from ethanol to get the pure compounds.
Tetraethyl [(pyridin-3-ylamino)methylene]bis(phosphonate) (4a, C14H26N2O6P2)
Yield 93% (white solid); m.p.: 154–156 °C; 1H NMR (400 MHz, CDCl3): δ = 8.21 (1H, s, Ar–H), 8.10 (1H, d, J = 8 Hz, Ar–H), 7.27 (1H, d, J = 8 Hz, Ar–H), 6.97 (1H, d, J = 8 Hz, Ar–H), 5.87 (1H, s, NH), 5.48–5.40 (1H, m, P–CH), 4.12–4.06 (8H, m, POCH 2CH3), 1.22–1.18 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 146.33, 142.48, 138.98, 126.34, 121.31, 64.04 (d, 1 J P–C = 11 Hz, POCH2CH3) 44.87 (d, 2 J P–C = 36 Hz, P–C–H), 16.35 (d, 3 J P–C = 28 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 21.16 ppm; IR (neat): \(\nu\) = 3217 (NH), 1260 (P=O), 1017 (P–O–C), 760 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 381 (48, [M + 1]+), 380 (35, [M]+), 355 (22), 346 (23), 152 (54), 91 (16), 90 (2), 77 (64).
Tetraethyl [(6-methylpyridin-2-ylamino)methylene]bis(phosphonate) (4b)
Yield 91% (white solid); physical data were found to agree with those described in Ref. [46].
Tetraethyl [(5-nitropyridin-2-ylamino)methylene]bis(phosphonate) (4c, C14H25N3O8P2)
Yield 90% (brown solid); m.p.: 147–149 °C; 1H NMR (400 MHz, CDCl3): δ = 8.93 (1H, s, Ar–H), 8.37 (1H, d, J = 8 Hz, Ar–H), 6.92 (1H, d, J = 4 Hz, Ar–H), 5.93 (1H, s, NH), 5.48–5.42 (1H, m, P–CH), 4.11–4.06 (8H, m, POCH 2CH3), 1.17–1.20 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 161.90, 148.82, 141.42, 136.57, 133.51, 107.76, 64.32 (d, 1 J P–C = 32 Hz, POCH2CH3), 54.87 (d, 2 J P–C = 36 Hz, P–C–H), 16.35 (d, 3 J P–C = 28 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 8.26 ppm; IR (neat): \(\nu\) = 3236 (NH), 1215 (P=O), 1018 (P–O–C), 723 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 426 (78, [M + 1]+), 425 (35, [M]+), 395 (43), 326 (22), 252 (51), 141 (18), 110 (8), 87 (34).
Tetraethyl [(5-chloropyridin-2-ylamino)methylene]bis(phosphonate) (4d, C14H25ClN2O6P2)
Yield 91% (white solid); m.p.: 185–187 °C; 1H NMR (400 MHz, CDCl3): δ = 8.31 (1H, s, Ar–H), 7.67 (1H, d, J = 8 Hz, Ar–H), 6.74 (1H, d, J = 4 Hz, Ar–H), 5.86 (1H, s, NH), 5.32–5.24 (1H, m, P–CH), 4.14–4.08 (8H, m, POCH 2CH3), 1.18–1.22 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 153.10, 146.62, 137.93, 126.48, 133.51, 117.76, 114.92, 64.06 (d, 1 J P–C = 32 Hz, POCH2CH3), 52.66 (d, 2 J P–C = 36 Hz, P–C–H), 16.32 (d, 3 J P–C = 28 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 18.58 ppm; IR (neat): \(\nu\) = 3248 (NH), 1247 (P=O), 1010 (P–O–C), 720 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 415 (84, [M + 1]+), 414 (65, [M]+), 395 (28), 366 (32), 315 (50), 291 (16), 262 (6), 97 (64).
Tetraethyl [(6-bromopyridin-2-ylamino)methylene]bis(phosphonate) (4e, C14H25BrN2O6P2)
Yield 88% (orange solid); m.p.: 133–135 °C; 1H NMR (400 MHz, CDCl3): δ = 7.58 (1H, t, 2 J = 16 Hz, Ar–H), 7.07 (1H, d, J = 8 Hz, Ar–H), 6.78 (1H, d, J = 8 Hz, Ar–H), 5.83 (1H, s, NH), 5.56–5.48 (1H, m, P–CH), 4.12–4.07 (8H, m, POCH 2CH3), 1.22–1.12 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 158.15, 143.40, 138.44, 117.6, 127.71, 116.01, 113.09, 106.07, 64.38 (d, 1 J P–C = 163.4 Hz, POCH2CH3), 54.86 (d, 2 J P–C = 36 Hz, P–C–H), 16.38 (d, 3 J P–C = 28 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 21.20 ppm; IR (neat): \(\nu\) = 3273 (NH), 1256 (P=O), 1021 (P–O–C), 735 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 460 (95, [M + 1]+), 459 (75, [M]+), 455 (22), 446 (63), 252 (34), 191 (26), 98 (2), 72 (44).
Tetraethyl [(4,6-dimethylpyrimidin-2-ylamino)methylene]bis(phosphonate) (4f, C15H29N3O6P2)
Yield 92% (white solid); m.p.: 156–158 °C; 1H NMR (400 MHz, CDCl3): δ = 6.74 (1H, s, Ar–H), 5.78 (1H, s, NH), 5.14–4.96 (1H, m, PCH), 4.11–4.06. (8H, m, POCH 2CH3), 2.58 (6H, s, 2 Ar-CH3), 1.22–1.14 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 164.89, 157.16, 115.49, 110.3, 107.2, 64.06 (d, 2 J P–C = 7.6 Hz, POCH 2CH3), 53.80 (d, 1 J P–C = 163.4 Hz, P–C–H), 24.75 (Ar-CH3), 16.35 (d, 3 J P–C = 7.0 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 20.85 ppm; IR (neat): \(\nu\) = 3248 (NH), 1245 (P=O), 1018 (P–O–C), 733 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 410 (84, [M + 1]+), 409 (65, [M]+), 385 (42), 346 (42), 252 (34), 168 (16), 90 (16), 67 (24).
Tetraethyl [(4-methoxy-6-methylpyrimidin-2-ylamino)methylene]bis(phosphonate) (4g, C15H29N3O7P2)
Yield 87% (pale yellow solid); m.p.: 166–168 °C; 1H NMR (400 MHz, CDCl3): δ = 6.12 (1H, s, Ar–H), 5.92 (1H, s, NH), 5.18–5.06 (1H, m, PCH), 4.14–4.09 (8H, m, POCH 2 CH3), 2.54 (3H,s, Ar–O-CH3), 1.48 (3H, s, Ar-CH3), 1.20–1.14 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 168.33, 166.51, 159.67, 110.69, 64.08 (d, 2 J P–C = 7.0 Hz, POCH 2CH3), 53.75 (Ar–O–CH3), 51.76 (d, 1 J P–C = 163.4 Hz, P–C–H), 24.86 (Ar–CH3), 16.35 (d, 3 J P–C = 7.0 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 17.95; IR (neat): \(\nu\) = 3267 (NH), 1234 (P=O), 1024 (P–O–C), 742 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 426 (68, [M + 1]+), 425 (42, [M]+), 385 (24), 346 (28), 242 (54), 176 (26), 98 (34), 64(16).
Tetraethyl [(4-chloro-6-methylpyrimidin-2-ylamino)methylene]bis(phosphonate) (4h, C14H26ClN3O6P2)
Yield 89% (orange solid); m.p.: 175–177 °C; 1H NMR (400 MHz, CDCl3): δ = 6.84 (1H, s, Ar–H), 5.74 (1H, s, NH), 5.14–5.02 (1H, m, PCH), 4.13–4.08 (8H, m, POCH2CH3), 2.57 (3H, s, Ar-CH3), 1.22–1.16 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.88, 162.58, 161.46, 112.31, 63.98 (d, 2 J P–C = 7.5 Hz, POCH 2 CH3), 52.78 (d, 1 J P–C = 18 Hz, P–C–H), 24.76 (Ar-CH3), 16.35 (d, 3 J P–C = 7.0 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 22.63 ppm; IR (neat): \(\nu\) = 3276 (NH), 1247 (P=O), 1016 (P–O–C), 728 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 430 (79, [M + 1]+), 429 (65, [M]+), 375 (28), 346 (32), 252 (45), 178 (16), 96 (21), 76 (46).
Tetraethyl [(5-bromopyrimidin-2-ylamino)methylene]bis(phosphonate) (4i, C13H24BrN3O6P2)
Yield 92% (white solid); m.p.: 140–142 °C; 1H NMR (400 MHz, CDCl3): δ = 8.65 (2H, s, Ar–H), 6.25 (1H, s, NH), 5.20–5.08 (1H, m, PCH), 4.15–4.07 (8H, m, POCH 2 CH3), 1.22–1.16 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 158.82, 156.3, 107.41, 64.36 (d, 2 J P–C = 7.0 Hz, POCH 2 CH3), 52.78 (d, 1 J P–C = 163.4 Hz, P–C–H), 16.36 (d, 3 J P–C = 7.5 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 21.50; IR (neat): \(\nu\) = 3248 (NH), 1245 (P=O), 1018 (P–O–C), 733 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 460 (82, [M + 1]+), 459 (68, [M]+), 425 (22), 386 (24), 256 (64), 190 (26), 145 (14), 82 (63).
Tetraethyl [(4-phenylpyrimidin-2-ylamino)methylene]bis(phosphonate) (4j, C19H29N3O6P2)
Yield 90% (yellow solid); m.p.: 149–151 °C; 1H NMR (400 MHz, CDCl3): δ = 8.14 (1H, d, J = 4.0 Hz, Ar–H), 7.66 (2H, d, J = 4.0 Hz, Ar–H), 7.46 (2H, d, J = 4.0 Hz, Ar–H), 7.38 (1H, t, 2 J = 8.0 Hz, Ar–H), 6.88 (1H, d, J = 4.0 Hz, Ar–H), 6.18 (1H, s, NH), 5.28–5.16 (1H, m, PCH), 4.15–4.10 (8H, m, POCH 2 CH3), 1.24–1.16 (12H, m, POCH2 CH 3) ppm; 13C NMR (100 MHz, CDCl3): δ = 159.93, 159.70, 159.14, 137.54, 130.44, 128.92, 128.11, 111.10, 62.38 (d, 2 J P–C = 7.0 Hz, POCH 2CH3), 52.56 (d, 1 J P–C = 18.5 Hz, P–C–H), 16.39 (d, 3 J P–C = 7.5 Hz, POCH2 CH 3 ) ppm; 31P NMR (161.7 MHz, CDCl3): δ = 20.88 ppm; IR (neat): \(\nu\) = 3296 (NH), 1263 (P=O), 1019 (P–O–C), 724 (P–C aliphatic) cm−1; ESI–MS: m/z (%) = 458 (84, [M + 1]+), 457 (45, [M]+), 411 (28), 346 (23), 277 (54), 162 (16), 98 (12), 84 (24).
In vitro cytotoxicity assay
All the synthesized compounds 4a–4j were tested for their cytotoxic activity using sulforhodamine-B (SRB) assay method [49]. MCF-7, DU-145, MG-63, HT-1080 and RPMI-8226 cancer cell lines were seeded for 24 h in a 96-well microtiter plates at a concentration of 1000–2000 cells/well and then the cells were incubated for 48 h with several concentrations of 100–6.25 µmol/cm3 (each repeated thrice). After treatment of 10% trichloroacetic acid, 150 mm3/well was added for 1 h at 4 °C, washed with distilled water for 3 times. Wells were stained with 0.4% (w/v) sulforhodamine-B 50 mm3/well for 10–30 min at room temperature. Unbound SRB was removed by washing with 1% acetic acid. The dye was solubilized with 100 mm3 of 10 mM Tris-base solution. The optical density (OD) of each well was measured spectrophotometrically with an ELISA microplate reader at 545 nm. To achieve the survival curve, the percent of surviving cells was calculated and plotted against different concentrations of the tested compounds. The IC50 values were calculated using sigmoidal concentration–response curve fitting models (Sigma plot software).
References
Dabholkar VV, Ansari FY (2010) Green Chem Lett Rev 3:245
Squarcialupi L, Colotta V, Catarzi D, Varano F, Betti M, Varani K, Vincenzi F, Borea PA, Porta N, Ciancetta A, Moro S (2014) Eur J Med Chem 84:614
Quiroga J, Diaz Y, Bueno J, Insuasty B, Abonia R, Ortiz A, Nogueras M, Cobo J (2014) Eur J Med Chem 74:216
Robinson SJ, Petzer JP, Terre’Blanche G, Petzer A, Walt MMV, Bergh JJ, Lourens ACU (2015) Eur J Med Chem 104:177
Chaubey A, Pandeya SN (2011) Asian J Pharm Clin Res 4:5
El-Hamid MKA, Mihovilovic MD, El-Nassan HB (2012) Eur J Med Chem 57:323
Tan Q, Zhang Z, Hui J, Zhao Y, Zhu L (2014) Bioorg Med Chem 22:358
Wan Z, Yao J, Tao Y, Mao T, Wang X, Lu Y, Wang H, Yin H, Wu Y, Chen F, Clercq ED, Daelemans D, Pannecouque C (2015) Eur J Med Chem 97:1
Baumann M, Baxendale IR (2013) Beilstein J Org Chem 9:2265
Lagoja IM (2005) Chem Biodiver 2:1
Clézardin P (2011) Bone 48:71
Stresing V, Daubine F, Benzaid I, Monkkonen H, Clezardin P (2007) Cancer Lett 257:16
Balakrishna A, Reddy MV, Rao PV, Kumar MA, Kumar BS, Nayak SK, Reddy CS (2011) Eur J Med Chem 46:1798
Shipman CM, Rogers MJ, Apperley JF, Russell RGG, Croucher PI (1997) Br J Haematol 98:665
Goldeman W, Goldeman AN (2014) Bioorg Med Chem Lett 24:3475
Santini D, Stumbo L, Spoto C, D’Onofrio L, Pantano F, Iuliani M, Fioramonti M, Zoccoli A, Ribelli G, Virzì V, Vincenzi B, Tonini G (2015) Breast Cancer Res 17:1
Reddy NB, Sundar CS, Krishna BS, Reddy KMK, Reddy CS (2014) Iranian J Org Chem 6:1227
Prasad SS, Jayaprakash SH, Syamasundar C, Sreelakshmi P, Bhuvaneswar C, Bhaskar BV, Rajendra W, Nayak SK, Reddy CS (2015) Phosphorus Sulfur Silicon Relat Elem 190:2040
Montalavetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R (2001) J Biol Chem 276:33930
Kotsikorou E, Song Y, Chan JMW, Faelens S, Tovian Z, Broderick E, Bakarala N, Docampo R, Oldfield E (2005) J Med Chem 48:6128
Paweł K, Barbara L, Giuseppe F (2000) Heteroat Chem 11:449
Lecercle D, Gabillet S, Gomis JM, Taran F (2008) Tetrahedron Lett 49:2083
Yokomatsu T, Yoshida Y, Nakabayashi N, Shibuya S (1994) J Org Chem 59:7562
Palacios F, Gil MJ, Marigorta EM, Rodriguez M (2000) Tetrahedron 56:6319
Lecercle D, Gabillet S, Gomis JM, Taran F (2008) Tetrahedron Lett 49:2083
Xia Y, Yang P, Sun Y, Wu Y, Mayers B, Gates B, Yin Y, Kin F, Yan H (2003) Adv Mater 15:353
Zhang M, Wang L, Ji H, Wu B, Zenge X (2007) J Nat Gas Chem 16:393
Beydoun D, Amal R, Low G, Mc Evoy S (1999) J Nanopart Res 1:439
Hosseini-Sarvari M, Etemad S (2008) Tetrahedron 64:5519
Hosseini-Sarvari M, Sharghi H, Etemad S (2008) Helv Chim Acta 91:715
Mirjafary Z, Saeidian H, Sadeghi A, Moghaddam FM (2008) Catal Commun 9:299
Kassaee MZ, Masrouri H, Movahedi F (2010) Monatsh Chem 141:317
Singh N, Mehra RM, Kapoor A (2011) J Nano-Eletron Phys 3:132
Seema R, Poonam S, Shishodia PK, Mehra RM (2008) Sol Energ Mat Sol Cells 92:1639
Thaslim Basha SK, Rasheed S, Sekhar KC, Raju CN, Ali MS, Rao CA (2014) Phosphorus Sulfur Silicon Relat Elem 189:1546
Kappe CO (2004) Angew Chem 43:6250
Kappe CO, Dallinger D (2006) Nat Rev Drug Disc 5:51
Jayaprakash SH, Krishna BS, Sundar CS, Prasad SS, Santhisudha S, Mohan G, Reddy CS (2016) J Heterocycl Chem 53:620
Reddy KMK, Santhisudha S, Mohan G, Peddanna K, Rao CA, Reddy CS (2016) Phosphorus. Sulfur Silicon Relat Elem 6:933
Mohan G, Santhisudha S, Reddy KMK, Reddy VN, Vijaya T, Reddy CS (2016) Heteroat Chem 27:269
Bacsa B, Bosze S, Kappe CO (2010) J Org Chem 75:2103
Duque JG, Pasquali M, Schmidt HK (2008) J Am Chem Soc 130:15340
Holtze C, Antonietti M, Tauer K (2006) Macromolecules 39:5720
Edwards WF, Young DD, Deiters A (2009) Org Biomol Chem 7:2506
Rahman KM, Thurston DE (2009) Chem Commun 20:2875
Reddy NB, Sundar CS, Krishna BS, Reddy KMK, Reddy CS (2014) Iranian J Org Chem 6:1227
Krutikov VI, Erkin AV, Pautov PA, Zolotukhina MM (2003) Russ J Gen Chem 73:187
Dąbrowska E, Burzyńska A, Mucha A, Matczak-Jon E, Sawka-Dobrowolska W, Berlicki Ł, Kafarski P (2009) J Organomet Chem 694:3806
Skehan P, Storeng R, Scudiero D, Monks A, McMahon I, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) J Nat Cancer Inst 82:1107
Rana K, Arora A, Bansal S, Chawla R (2014) Indian J Pharm Sci 76:339
Shamsuzzaman, Ayaz MD, Hena K, Gatoo MA (2014) Arabian J Chem 7:461
Prakash SS, Poonam SS, Sarav AD, Maulik PS (2011) Ann Bio Res 2:51
Acknowledgements
The authors render their sincere thanks to Prof. C. Devendranath Reddy for his helpful discussions, and they are also grateful to DST-SERB, New Delhi, India for providing financial support through Project No. SB/S1/OC-96/2013, Dt. 05-11-2014 and anticancer drug screening facility in ACTREC, Tata Memorial Centre, Navi Mumbai for in vitro SRB assay of compounds for anticancer activity.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mohan, G., Santhisudha, S., Reddy, N.M. et al. Nano ZnO catalyzed green synthesis and cytotoxic assay of pyridinyl and pyrimidinyl bisphosphonates. Monatsh Chem 148, 1843–1851 (2017). https://doi.org/10.1007/s00706-017-2000-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-017-2000-2