
Abstract
Canine parvovirus (CPV) infection causes severe gastroenteritis in canines, with high mortality in puppies. This virus evolved from feline panleukopenia virus by altering its transferrin receptor (TfR), followed by the emergence of CPV-2 variants in subsequent years with altered immunodominant amino acid residues in the VP2 protein. While previous studies have focused on the VP2 gene, there have been fewer studies on non-structural protein (NS1 and NS2) genes. In the present study, CPV genome sequences from clinical samples collected from canines throughout India in 2023, previous Indian CPV isolates from 2009–2019, and the current Indian CPV vaccine strain were compared. The study showed that the CPV-2c (N426E) variant had almost completely replaced the previously dominant CPV-2a variant (N426) in India. The Q370R mutation of VP2 was the most common change in the recent CPV-2c strain (CPV-2c 370Arg variant). Phylogenetic analysis showed the existence of three clades among the recent CPV-2c strains, and sequence analysis identified several new sites of positive selection in the VP1 (N-terminus), VP2, NS1, and NS2 protein-encoding genes in recent CPV strains, indicating the emergence of new CPV-2c variants with varied antigenic and replication properties. The predominant ‘CPV-2c 370Arg variants’ were grouped with the Chinese and Nigerian CPV-2c strains but were separate from the CPV vaccine strain and earlier isolates from our repository. VP2 epitope analysis predicted nine amino acid variations (including two new variations) in four potential linear B-cell epitopes in the CPV-2c 370Arg variants that might make vaccine failure more likely. This pan-Indian study lays the foundation for further research concerning the dynamics of virus evolution and understanding genetic mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Canine parvovirus 2 (CPV-2) is a member of the genus Protoparvovirus of the family Parvoviridae [1] that causes acute hemorrhagic gastroenteritis and myocarditis in wild and domestic canines [2]. The disease is responsible for a significant number of animal deaths, particularly in puppies under six months of age. Clinical signs of the disease include vomiting, fever, diarrhoea (often bloody), hemorrhagic enteritis, myocarditis, and leukopenia [3]. CPV-2 is a non-enveloped virus with a diameter of about 25 nm, and its genome is a single-stranded linear DNA molecule of 5 kb that contains two open reading frames [4], the first of which encodes the structural proteins VP1 and VP2 and the second of which encodes the non-structural proteins NS1 and NS2 [5].
In the late 1970s, CPV-2 emerged as a novel canine pathogen and spread rapidly throughout the world. This virus evolved from feline panleukopenia virus (FPV) through interspecies transmission with other carnivores such as minks and foxes, eventually infecting dogs. Therefore, CPV-2 can be regarded as a variant of FPV that specifically affects canines [6]. After its emergence, CPV-2 underwent genetic and antigenic changes within a few years, leading to the evolution of newer strains, named CPV-2a and CPV-2b [7], which differ at amino acid residue 426 of the VP2 gene, which is asparagine in CPV-2a and aspartic acid in CPV-2b. Later, an additional amino acid variation was documented in CPV-2a and CPV-2b isolates in Germany. These strains, which have a Ser→Ala substitution at amino acid residue 297 of VP2, were designated as ‘New CPV-2a/2b’ in the early 1990s and now as ‘CPV-2a/2b variants’ [8]. In 2000, a new antigenic variant with glutamic acid at amino acid residue 426 of the VP2 gene was reported in Italy and was subsequently designated as ‘CPV-2c’ [9]. Thus, CPV strains are differentiated based on the variations observed at amino acid residues 297 and 426 of the VP2 capsid protein [10]. Currently, New CPV-2a (CPV-2a variant), New CPV-2b (CPV-2b variant), and CPV-2c appear to have replaced the prototype CPV-2a/2b and become the predominant types co-circulating in many Asian countries [5, 11,12,13,14]. The CPV-2 prototype is no longer found in canines but is still used in CPV vaccines [15]. Previous studies have shown that the CPV-2a variant is the predominant strain in India, but there have been a few reports of CPV-2b and CPV-2c [11, 12, 16,17,18,19,20]. However, recent studies in neighbouring countries, including China, Taiwan, Myanmar, and Thailand have found a higher occurrence of CPV-2c, increasing the risk that this variant will continue to spread [21,22,23].
So far, there has not been a comprehensive molecular surveillance study of CPV encompassing all of the geographical areas of India. The majority of the studies have focused on the VP2 capsid protein gene, but research on the non-structural genes NS1 and NS2 and the gene encoding VP1, which is important for viral replication and apoptosis, also needs to be undertaken. It is crucial to analyze the complete genome of the virus over time in a large geographical region for a better understanding of its evolution and spread. Therefore, the aim of this study was to compare the genomic profiles of CPV strains from 2009–2019 to those of current CPV strains distributed across all six geographical regions of India.
Materials and methods
Clinical samples
A total of 203 faecal samples/rectal swabs were collected from dogs with suspected CPV infection from different locations covering all six geographical regions of India during 2023, as detailed in Table 1. The samples were resuspended in 0.1 M phosphate-buffered saline (pH 7.4) and transported to the laboratory on ice for processing. After centrifugation at 6000 rpm for 15 min at 4°C, the supernatant was collected and stored at -40°C for further use. In addition to the clinical samples, repository isolates of canine parvovirus that had been identified and deposited in the collection of the Department of Veterinary Microbiology, Rajiv Gandhi Institute of Veterinary Education and Research (RIVER), India, over a period of time (2009–2019) (Supplementary Table S1) were also used in this study, together with the current CPV vaccine virus.
Extraction of viral DNA
A 200-µL aliquot each of processed sample was treated with a mixture containing lysis buffer, proteinase K, and magnetic beads and kept at 56°C for 5 min. The viral DNA was extracted from clinical samples, repository isolates, and the vaccine, using a Nucleic Acid Extraction Kit (Zybio Inc. Chongqing, China) according to the manufacturer's protocol, and stored at -40 °C until further use.
PCR amplification
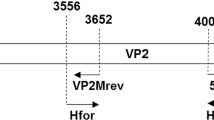
Samples were screened for the presence of CPV using the primer pair Hfor (5’-CAGGTGATGAATTTGCTACA-3') and Hrev (5’-CATTTGGATAAACTGGTGGT-3') to amplify a 630-base-pair (bp) fragment of the CPV VP2 gene encoding amino acid residues at positions 297, 300, 305, 375, and 426, which are significant for CPV strain differentiation [9]. The PCR reaction mixture consisted of a 1X concentration of Ampliqon AQ97 DNA Polymerase Master Mix, Hfor/Hrev primers (0.2 µM each), 2 µL of template DNA, and nuclease-free water to make up a final volume of 50 µL. The conditions for PCR amplification were described previously by Karanam et al. [20]. The PCR-amplified products were resolved on a 1.5% agarose gel in Tris-acetate EDTA buffer and visualized using a UV transilluminator (Syngene, Norway).
CPV strain identification
Seventy-four amplified PCR products of recent clinical samples were selected, representing all six geographical regions of India (Table 1). The PCR products were extracted from a gel using a QIAGEN Gel Extraction Kit and sent for custom sequencing in both directions (5'–3' and 3'–5') using an Applied Biosystems 3100 automated sequencer. The specificity of the sequences was checked using the nucleotide Basic Local Alignment Search Tool (BLAST) on the NCBI website (http://www.ncbi.nlm.nih.gov), and the nucleotide sequences were aligned with the corresponding CPV sequences available in the GenBank database using the Clustal Omega program of the MEGA 11 software package [24]. The partial sequences of the VP2 gene were submitted to the GenBank database (www.ncbi.nlm.nih.gov/genebank) under the accession numbers OR339704 to OR339759 (Table 1). The partial VP2 sequences of the 74 recent CPV isolates, 21 repository isolates (2009–2019 isolates) from our laboratory (Supplementary Table 1), seven CPV vaccine strains, and 22 reference isolates from various parts of the world (NCBI, GenBank) were used to construct a phylogenetic tree (outgroup rooted) by the neighbor-joining method, using the MEGA 11 program [24].
Whole-genome sequencing
Whole-genome sequencing was done for molecular characterization of the structural (VP1 and VP2) and non-structural (NS1 and NS2) protein genes of CPV. Eighteen randomly selected representative recent samples that tested positive using the primer pair Hfor/Hrev, obtained from diverse locations in India (three samples from each region), 11 repository CPV isolates (2009–2019 isolates), and a current Indian CPV vaccine strain, were subjected to PCR amplification to yield seven overlapping PCR products for generation of the complete CPV genome sequence, using the seven sets of primers listed in Supplementary Table S2, followed by custom sequencing and sequence alignment. The PCR reaction mixture and the conditions of the PCR assay to amplify each fragment were carried out as described by Karanam et al. [20]. The details of the 18 clinical samples and 11 repository CPV isolates used for whole-genome sequence analysis are shown in Table 1 and Supplementary Table S1, respectively.
Sequencing and sequence analysis
The seven overlapping amplified PCR products of CPV sequences from each sample were subjected to gel extraction and custom sequenced as described above, using the seven sets of sequencing primers listed in Supplementary Table S2. The sequencing results were checked by BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and aligned. The coding sequences of VP1 (N-terminus), VP2, NS1, and NS2 were aligned separately with the corresponding complete coding sequences of the repository isolates, Indian CPV vaccine strain, and reference CPV strains available in GenBank, using the Clustal Omega program of the MEGA 11 software package [24] to identify new substitutions and mutations. Amino acid positions 297 and 426 of VP2 were examined to identify strains [13, 25, 26], and 12 other amino acid positions in VP2 (5, 87, 267, 300, 322, 323, 324, 334, 341, 370, 440, and 555) that have been reported to affect infectivity and pathogenicity [13, 25,26,27,28] were also examined for mutations or variations.
Phylogenetic analysis based on the VP2 and NS1 genes
Phylogenetic analysis was performed separately based on the complete coding sequences of the VP2 and NS1 genes. The phylogenetic tree (outgroup-rooted) was constructed by the neighbor-joining method, using the MEGA 11 program [24], using the sequences of the 18 recent clinical samples and the corresponding sequences of repository isolates, the Indian vaccine strain, and reference CPV strains from various parts of the world available in the NCBI nucleotide database. The confidence level of branching in the phylogenetic tree was evaluated as described by Karanam et al. [20].
Recombination analysis
GARD (Genetic Algorithms for Recombination Detection) and RDP4 analysis (http://www.datamonkey.org) were performed to identify breakpoints and recombination events in the VP1, VP2, NS1, and NS2 genes of the CPV strains as described by Karanam et al. [20].
Selective pressure analysis
Selective pressure was evaluated based on the ratios of non-synonymous (dN) to synonymous (dS) mutations (dN/dS) within the VP1,VP2, and NS1 genes of CPV strains, using the single-likelihood ancestor counting (SLAC) method (http://www.datamonkey.org) as described by Karanam et al. [20]. The dN–dS ratio <1.0, = 1.0, and >1.0 indicate negative, neutral, and positive selection, respectively. Statistical analysis was performed to compare the targeted genes of the recent CPV strains and repository isolates, using the chi-square test of independence (χ2 test) in SPSS software version 20. A χ2 cal value that is higher than the χ2 tab value indicates that the null hypothesis (H0) is rejected at a 5% significance level.
B cell epitope prediction
An effective vaccine should stimulate cellular and humoral immune responses against the target pathogen. Hence, we also sought to identify the potential B cell epitopes from our study sequences. We limited our analysis to the structural protein of the CPV (VP2 gene), as these are the most accessible antigens for engaging B cell receptors. The Immune Epitope Database (IEDB), a sequence-based tool, was used to identify potential linear B-cell epitopes using the Bepipred 2.0 algorithm.
Results
Out of 203 clinical samples, 162 (79.8%) were found to be positive for the presence of CPV by PCR, using the primers Hfor/Hrev, which target a portion of the VP2 gene, yielding a specific amplicon of 630 bp. Details about the samples collected in India and their PCR results are shown in Table 1.
Identification of CPV strains
Partial VP2 sequences of 74 recently collected clinical samples that tested positive by PCR were found to be completely identical to those of other CPV strains identified by BLAST analysis of the NCBI GenBank database. Of the samples tested, 59 (79.7%) were identified as CPV-2c (297Ala, 426Glu), 14 (18.9%) were identified as CPV-2a (297Ala, 426Asn), and only one (1.3%) was identified as CPV-2b (297Ala, 426Asp) (Fig. 1, Table 1, and Supplementary Table S3).

CPV strain identification in recently collected CPV samples covering wide geographical areas in India. The CPV-2c strain was predominant in most parts of India, with few co-circulating CPV-2a variants and a lone CPV-2b variant
Four variable amino acid sites were identified in recent CPV strains when compared to the repository CPV isolates and the Indian vaccine strain (Supplementary Table S3). Notably, the Gln370→Arg mutation was detected in 61 (82%) of the 74 recent CPV isolates studied. The majority of recent CPV-2c isolates (55/59, 93.2%) had the Gln370→Arg substitution (CPV-2c 370Arg variant), but the recent CPV-2a variant (6/14, 42.8%) (CPV-2a 370Arg variant) was also detected. The lone CPV-2b variant did not have the Gln370→Arg variation. The other three variations included Gly286→Ser in one CPV-2c isolate (OR296260), Glu368→Lys in six CPV-2a/2c isolates (OR339747, OR339748, OR339749, OR339750, OR339753, OR339720), and Asp373→Asn in three CPV-2a isolates (OR339749, OR339751, OR339753). The previously predominant variation at amino acid residue 440 (Thr→Ala) that was present in our earlier repository isolates (CPV-2a variant) was not observed in most of our recent CPV isolates, especially the recent CPV-2c isolates.
Phylogenetic analysis based on partial VP2 gene sequences
Phylogenetic analysis was performed using 74 recent CPV isolates, 21 repository isolates (2009–2019) from our laboratory, seven CPV vaccine strains, and 22 reference CPV sequences representing various strains from various parts of the world (NCBI, GenBank) (Fig. 2). The recent CPV isolates analyzed in this study formed three clades (clades I, II, and III). Most of the recent CPV-2c isolates with an arginine substitution at position 370 (CPV-2c 370Arg variant) formed a monophyletic clade (clade I), together with Chinese CPV-2c strains, that was separate from the vaccine strains. The recent CPV-2a variants and CPV-2b variant formed a separate monophyletic clade (clade II) together with Chinese CPV-2a strains and the 2009–2019 repository isolates (CPV-2a/2b variant), but a few CPV-2c strains (OR339717, OR339736, OR296269, and OR339719) without a change at amino acid position 370 (Gln370) also were clustered in clade II. Similarly, some of the CPV-2c strains (10 sequences) without a change at amino acid position 324 (Tyr324) grouped with the prototype CPV-2c strain and the vaccine strains and formed a separate clade (clade III). A phylogenetic tree constructed using the maximum-likelihood method showed a similar distribution pattern of the CPV strains (data not shown).

Phylogenetic tree based on partial nucleotide sequences of the VP2 gene, depicting the phylogenetic relationship among parvovirus strains. The tree was constructed in MEGA version 11.0 by the neighbor-joining method with the Kimura 2-parameter model and 1000 bootstrap replications. Recent parvovirus strains sequenced in this study (indicated by solid circles) were analyzed together with repository isolates (2009–2019) (indicated by solid triangles), CPV vaccine strains (indicated by solid squares), and reference sequences representing CPV strains from various parts of the world
Whole-genome analysis
The complete genome sequences (4,363 nt) of 30 CPV sequences analyzed in this study (including 18 recent CPV isolatates, 11 repository CPV isolates, and one Indian CPV vaccine strain) were submitted to the GenBank database (www.ncbi.nlm.nih.gov/genebank) under the accession numbers OR296260 to OR296277 (18 recent CPV isolates), OR296278 to OR296288 (11 repository CPV isolates), and OR296289 (CPV vaccine strain) (Table 1 and Supplementary Table S1). BLAST analysis revealed that the study sequences had 98.9 to 100% nucleotide sequence identity to other CPV isolates and contained four putative open reading frames (ORFs) (NS1, NS2, VP1, and VP2).
Analysis of the N-terminal amino acid sequence of the VP1 gene
VP1 consists of the entire VP2 sequence plus an extra, unique N-terminal sequence. The variations in the VP1 N-terminal amino acid sequence are shown in Supplementary Tables S4 and S5. The VP1 gene is 2184 nt long with no insertions or deletions, encoding a protein of 728 amino acids. Of the 11 repository isolates from 2009 to 2019, one had a Ser25Ile substitution, one had a Gly82Glu substitution, and three had a Leu95Val substitution in VP1 with respect to the CPV vaccine strain. However, in the CPV strains from 2023, an additional 12 amino acid substitutions were identified in VP1: Glu30Lys, Ala37Pro, Asp43Asn, Glu44Lys, Ala45Ser, Ala47Pro, Asp64Asn, Leu83His, Tyr84Phe, Arg87Lys, Lys116Thr, and His120Leu. The Leu83→His mutation, which was not present in earlier isolates, was found in 83.3% of the recent CPV isolates. In addition, there was an increase in variations at amino acid positions 25 (Ser→Ile) and 95 (Leu→Val) in recent CPV strains.
Sequence analysis of the VP2 gene
Most of the viral capsid is composed of VP2, with VP1 comprising a smaller proportion. The sequencing results showed that the VP2 gene is 1755 nt long, with no insertions or deletions, encoding a protein of 585 amino acids. The variations in the VP2 protein found in this study are shown in Table 2 and Supplementary Table S6. The repository isolates had variations at 11 amino acid positions – 101, 219, 267, 297, 300, 305, 324, 375, 386, 401, and 440 – when compared to the Indian CPV vaccine strain. In the recent CPV strains, an additional five new mutations were identified in the VP2 protein: Ala189Ser (OR296260), Met190Ile (OR296277), Gly286Ser (OR296260), Gln370Arg (OR296260-OR296277), and Lys582Asn (OR296260). All 18 of the recent CPV isolates had the Asn426→Glu substitution, which is characteristic of CPV-2c strains.
Sequence analysis of the NS1 gene
The sequencing results showed that the NS1 gene is 2007 nt long, encoding a protein of 669 amino acids, with no insertions or deletions in the coding region. The variations in the NS1 protein found in this study are shown in Table 3 and Supplementary Table S7. The repository isolates had three variations at amino acid positions 19, 207, and 237 with respect to the CPV vaccine strain. The Lys19Arg substitution was present in 91% of the repository CPV-2a variants but was observed in only one recent CPV strain. In the recent CPV samples, an additional 23 new mutations were identified in the NS1 protein at amino acid positions 60, 217, 229, 231, 242, 247, 260, 263, 351, 383, 389, 392, 427, 544, 545, 554, 572, 583, 584, 586, 596, 630, and 664. In the recent CPV-2c isolates, 83.3% (15 of 18 samples) had a variation at amino acid position 583 (Lys→Glu), and 72.2% (13 of 18 samples) had the substitutions Ile60Val and Leu630Pro, which were not observed in our repository isolates but have been observed in CPV strains circulating in other countries.
Sequence analysis of the NS2 gene
The NS2 gene is 498 nt in length, encoding a protein of 166 amino acids. Variations were observed at amino acid positions 19 and 110 in the repository isolates compared to the vaccine strain (Supplementary Tables S8 and S9). In addition, four new amino acid substitutions were found in the recent CPV strains: Ile60Val, Lys92Arg, Asp93Glu, and Thr94Ala. Among the recent CPV-2c isolates, 83.3% (15 of 18 samples) had a variation at amino acid position 92, and 72.2% (13 of 18 samples) had a variation at amino acid position 60, neither of which had been observed in the repositories.
Phylogenetic analysis based on the complete VP2 gene
Phylogenetic analysis based on the complete VP2 gene (1755 nt) was performed using sequences of 18 recent CPV isolates, 21 repository isolates (2009–19) from our laboratory, six CPV vaccine strains, and 29 reference strains from different parts of the world (NCBI, GenBank) (Fig. 3). All of the recent CPV-2c isolates formed a separate monophyletic cluster (clade I) together with CPV- 2c sequences from China, Nigeria, and north-eastern India. All of our repository CPV-2a/2b/2c isolates (2009–2019) belonged to clade II, in which almost all of the CPV-2a variant strains clustered together (subclade IIa) with CPV-2a isolates from China, and few repository isolates (CPV-2a/2b/2c) were clustered with CPV-2a/2b/2c strains from Italy, Brazil, and the USA in subclade IIb. However, the CPV vaccine strains (CPV-2) were separated from the recent CPV sequences and instead clustered with the prototype CPV/FPV strains (clade III). A similar distribution pattern was observed in a phylogenetic tree constructed using the maximum-likelihood method (data not shown).

Phylogenetic tree based on complete nucleotide sequences of the VP2 gene, depicting the phylogenetic relationship among parvovirus strains. The tree was constructed in MEGA version 11.0 by the neighbor-joining method with the Kimura 2-parameter model and 1000 bootstrap replications. Recent parvovirus strains sequenced in this study (indicated by solid circles) were analyzed together with repository isolates (2009–2019) (indicated by solid triangles), CPV vaccine strains (indicated by solid squares), and reference sequences representing CPV strains from various parts of the world
Phylogenetic analysis based on the NS1 gene
Phylogenetic analysis based on the complete NS1 gene (2007 nt) was performed using sequences of 18 recent CPV isolates, 11 repository isolates (2009–2019) from our laboratory, one CPV vaccine strain, and 17 reference strains from various parts of the world (NCBI, GenBank) (Fig. 4). The majority of the recent CPV-2c isolates (13 sequences) had a substitution at amino acid residue 60 (Ile→Val) and formed a monophyletic clade (clade II) together with CPV-2c strains from China and Nigeria, as well as one strain from India (Mizoram), that grouped separately from the CPV vaccine strain and the CPV repository isolates (subclade Ia). A few recent CPV-2c isolates (OR296261, OR296263, OR296264, OR296273, and OR296275), which lacked a mutation at amino acid residue 60 (Ile60) clustered with the prototype CPV/FPV strains in subclade Ib. A phylogenetic tree constructed using the maximum-likelihood method showed a similar pattern of distribution of CPV strains (data not shown).

Phylogenetic tree based on complete nucleotide sequences of the NS1 gene, depicting the phylogenetic relationship among parvovirus strains. The tree was constructed in MEGA version 11.0 by the neighbor-joining method with the Kimura 2-parameter model and 1000 bootstrap replications. Recent parvovirus strains sequenced in this study (indicated by solid circles) were analyzed together with repository isolates (2009–2019) (indicated by solid triangles), CPV vaccine strain (indicated by solid squares), and reference sequences representing CPV strains from various parts of the world
Recombination analysis
RDP4 analysis did not identify any breakpoints in the CPV sequences from this study, indicating that no recombination events had occurred.
Selective pressure analysis of the VP1-VP2-coding region
In the repository isolates (2009–2019), it was found that the overall dN-dS substitution rate was 0.106, which indicates negative selection (dN/dS < 1.0). However, sites exhibiting positive selection were identified at amino acid residues 25, 82, and 95 in the N-terminus of VP1 and at residues 101, 219, 267, 297, 300, 305, 324, 375, 386, 401, and 440 in VP2. In recent strains of CPV, the overall dN-dS substitution rate was found to be 0.212, which is also less than 1.0. Additional positively selected sites were identified at amino acid positions 30, 37, 43, 44, 45, 47, 64, 83, 84, 87, 116, and 120 in the N-terminus of VP1 and 189, 190, 286, 370, 426, and 582 in VP2 (Supplementary Fig. S1). The differences in the substitution rates in the VP1-VP2-coding region between the recent CPV strains and repository isolates were found to be statistically significant (p < 0.05).
Selective pressure analysis of the NS1-coding region
In the repository isolates (2009–2019), the overall dN-dS substitution rate was found to be 0.06, indicating negative selection (dN/dS < 1.0), but sites exhibiting positive selection were identified at amino acid positions 19, 207, and 237 in the NS1 protein. In the recent CPV strains, the overall dN-dS substitution rate was found to be 0.246 (dN/dS < 1.0), and additional positively selected sites were observed at amino acid positions 60, 217, 229, 231, 242, 247, 260, 263, 351, 383, 389, 392, 427, 544, 545, 554, 572, 583, 584, 586, 596, 630, and 664 in NS1 (Supplementary Fig. S2). The difference in the substitution rates in the NS1-coding region between the recent CPV strains and repository isolates were found to be statistically significant (p < 0.05).
Prediction of B cell epitopes
We identified 19 predicted peptide epitopes in the CPV VP2 capsid protein of the CPV vaccine strain, repository isolates, and recent CPV sequences (Supplementary Tables S10, S11, and S12). These epitopes were located between amino acids 4 and 580 (Supplementary Table S10). In the repository isolates (2009–2019), seven amino acid variations were identified in the four major peptide epitopes compared to the CPV vaccine strains (Supplementary Table S11). However, most of the recent CPV strains had two additional variations at amino acid residues 370 and 426 in the major peptide epitope region (Supplementary Table S12).
Discussion
In this study, a PCR assay was conducted to screen the samples for CPV, and the overall positivity rate was 79.8%. Similar results were also obtained in our earlier studies [11, 12] and studies by other researchers in India [17, 29]. The majority of the new CPV isolates were CPV-2c, with the CPV-2a variant found in a few samples, and the CPV-2b variant found in only one sample. All previous reports [12, 18, 30,31,32] have shown CPV-2a to be the predominantly circulating CPV variant in India, but a few studies indicated the presence of CPV-2b/2c in certain isolated pockets in small numbers [18, 33, 34]. The present study is the first of its kind in which CPV-2c was found to be the predominant circulating variant in all of the geo-climatic zones of India and has almost completely replaced the previously predominant CPV-2a variant. The CPV-2b variant was found to be almost extinct in the present study. Fu et al. [35] reported that the prevalence of CPV-2c in China increased annually beginning in 2017 and became more prevalent than the CPV-2a variant strain in 2018, suggesting that CPV-2c has become the primary CPV variant in China. Hao et al. [36] also found that CPV-2c was replacing CPV-2a as the new dominant variant in Asia, South America, North America, and Africa. CPV-2c has completely replaced the previously dominant CPV-2a variant in the North-East and Eastern regions, and almost completely in the Central region of India. Only a few CPV-2a variants and a lone CPV-2b variant were detected in the North, West, and Southern regions. This observation suggests that the current CPV-2c variant in India might have originated in neighboring Asian countries that share borders with states in the North-Eastern zone of India and entered through their very porous borders.
In the immunodominant VP2 protein, the variation at amino acid position 370 (Gln→Arg) was found in the majority of the recent CPV-2c strains and a few CPV-2a strains, so almost all of the recent CPV-2c strains in this study were considered ‘CPV-2c 370Arg variants’. This variation was not observed in the repository isolates (2009–2019) identified from various regions of India. Guo et al. [37] speculated that residue 370 is close to residues 375 and 377 and is associated with the ability of CPV to hemagglutinate or alter the pH dependence of hemagglutination. Similarly, residue 370 is close to residues 379 and 384, and changes at this position might affect binding to the canine transferrin receptor (Tfr), thereby affecting the host range of the virus. Substituting Gln370 with Arg might result in a conformational change or affect receptor binding through neighboring residues. Most of our recent CPV-2c strains had the amino acid substitutions Phe267Tyr, Tyr324Ile, and Gln370Arg, which are characteristic of CPV-2c strains circulating in Asian countries, including Laos [38], Taiwan [39], South Korea [40], and China [35, 41]. Another variation, Ala5→Gly, which has been reported in CPV-2c strains from many Asian countries [42,43,44,45], was not observed in recent CPV strains or repository isolates from India. This indicates that this mutation may not have a single origin but has evolved independently due to the increasing selective pressure at this site, as this mutation was not present in some variants from China [41].
We identified two unique mutations that had not been reported previously: Gly286→Ser (in a CPV-2c strain) and Glu368→Lys (in CPV-2a/2c strains). These mutations are in the GH loop of the VP2 protein (residues 267–498, located between the bG and bH strands), and the most significant variability was seen in this antigenic region [46]. Thus, the identification of such unique mutations in the present study indicated that the CPV strains are under constant selection pressure and are constantly mutating, leading to the evolution of new CPV types or variants [12]. Another unique amino acid variation was identified at amino acid position 373, Asp→Asn (in a CPV-2a variant), located within the VP2 flexible loop, a pH-sensitive structure that governs binding to divalent ions in FPV and CPV-2 [10]. The Tyr324→Ile substitution was identified in most CPV-2c and CPV-2a variants and the lone CPV-2b variant. This mutation was widely reported earlier in India [11, 12, 18], in Asian countries [26, 47,48,49,50], and in Italy [51]. Notably, some recent CPV-2c strains in this study were observed to lack a variation at the amino acid residue 324 and therefore resembled the prototype (Tyr324). Since the Tyr324→Ile mutation is located near residue 323 in the VP2 protein, which participates in the recognition of Tfr type I [52], the variation at 324 residues is likely to have an effect on the host range of CPV.
In our recent CPV samples (CPV-2c 370Arg variants), we did not observe the previously predominant variation at amino acid residue 440 (i.e., Thr→Ala). Similarly, a few recent CPV-2a variants with the Gln370Arg variation are also observed with the prototype Thr440. In contrast, the repository CPV-2a/2b variants without the Gln370Arg substitution had the Thr440→Ala substitution, as also reported by other researchers [53, 54]. The amino acid residue 440 of VP2 is located on the top of the 3-fold spike of loop 4 and might be a variable site [55]. The frequency of Thr440→Ala mutations peaked in 2014 [27, 36, 54, 56], and this mutation was not identified in the recent CPV strains. Therefore, all of the recent CPV strains with the Gln370Arg variation had the prototype Thr440. This distinction in the amino acid sequence at these positions potentially aids in finer differentiation between various CPV-2 genetic variants.
In addition, the recent CPV strains had ??four?? additional amino acid variations in comparison to the vaccine strain at positions 189, 190, 286, and 582, which were neither observed in our repository isolates nor reported in earlier CPV strains studied. These four unique single mutations are not located within the VP2 protein loops, and their effect on the biological functions of CPV requires further investigation.
Phylogenetic analysis based on partial VP2 gene sequences revealed the existence of three clades of CPV-2c strains that are currently co-circulating in the canine population in India, together with a few CPV-2a/2b variants. Most of the CPV-2c strains, i.e., the CPV-2c 370Arg variants, clustered with Chinese CPV-2c isolates and a few Indian CPV-2c strains from the North-East region. The clustering with the Chinese CPV-2c strains suggests that the “Asian CPV-2c” lineage had progressively replaced other lineages and become the dominant strain in several areas of Asia within a few years [41]. The fact that this cluster included CPV-2c strains from the North-Eastern states of India indicated a possible potential link between the CPV-2c variants in India and the Chinese CPV-2c isolates, with North-Eastern states of India, with their porous borders possibly serving as entry points. Notably, this group of CPV-2c strains was phylogenetically separate from the current Indian CPV vaccine strains. In clade II, very few recent CPV-2c strains without a variation at position 370 were grouped along with recent CPV-2a/2b strains, repository isolates (CPV-2a/2b variant), and the Chinese CPV-2a variants. This is in agreement with earlier findings [18, 29]. Some recent CPV-2c strains without a variation at residue 324, with the prototype Tyr324, formed a separate clade (clade III) together with the prototype CPV-2c strain and vaccine strains. These findings provide valuable insights into the genetic relationships and potential transmission dynamics of CPV variants within India and globally. The clustering patterns highlight the possible origins and divergence of CPV strains, underscoring their rapid evolution.
Phylogenetic analysis based on complete VP2 gene sequences showed that all of the recent CPV-2c strains grouped in a clade with Chinese, Nigerian, and North-East Indian CPV-2c strains. These recent CPV-2c strains clustered separately from the repository CPV-2a/2b/2c isolates (2009–2019), prototype Italian CPV-2c isolates, and current Indian CPV vaccine strains.
VP1 consists of the whole sequence of VP2 plus an extra, unique N-terminal sequence. In addition to the three variation sites observed in our repository isolates (2009–2019), recent CPV strains (2023) had 12 additional amino acid variation sites in the VP1 protein (N-terminus) in comparison to the vaccine strain. The substitution Leu83→His was observed in the majority of the recent CPV-2c strains but was not observed in our repository isolates or reported in any Indian CPV strains analyzed earlier, although this variation has been observed in CPV strains from China, the USA, and Nigeria. The VP1 N-terminal sequence is essential for virus-cell interaction because it has phospholipase A2 enzyme activity that is necessary for viral infectivity [57], as well as a potential nuclear transport sequence [58]. Therefore, variations in the N-terminal VP1 sequence can potentially affect viral infectivity.
There have been no detailed studies on the non-structural genes of CPV strains from India. These genes are associated with the replication of the CPV genome. In the NS1 protein, in addition to three variations observed in the repository isolates (2009–2019), recent CPV strains (2023) were found to have 23 additional amino acid substitutions when compared with the vaccine strain. Notably, the unique mutations Ile60Val and Leu630Pro were observed in the majority of the recent CPV-2c isolates but were not found in our repository isolates. The NS1 protein of Asian-origin CPV-2a variant and CPV-2c strains frequently contained Ile60→Val, Phe544, Val545, and Lys630→Pro. These variations are characteristic of Asian-origin CPV-2c strains [35, 54, 59]. Amino acid position 60 is located in the N-terminal portion of the replication domain. The Leu630→Pro mutation, which is located in the C-terminal transactivation domain, may be responsible for local adaptation, as suggested by Han et al. [13]. Similarly, another unique variation at residue 583 (Lys→Glu) of the NS1 protein was observed in the majority of the recent CPV-2c isolates but was not observed in our repository isolates (2009–2019). This mutation was identified in earlier CPV strains circulating in other countries, including in CPV-2b isolates [60].
Phylogenetic analysis based on the complete NS1 gene revealed that the majority of recent CPV-2c isolates with a substitution at residue 60 were clustered in clade II with Chinese, Nigerian, and North-East Indian CPV-2c strains and grouped separately from the CPV vaccine strain and strains from the repository. However, a few recent CPV-2c isolates without a substitution at amino acid position 60 (Ile60) clustered with the prototype CPV/FPV strains.
In the NS2 protein, in addition to the two variations observed in the repository isolates (2009–2019), recent CPV strains (2023) had four additional amino acid variations when compared with the vaccine strain. The majority of the recent CPV-2c isolates had variations at residues 60 and 92, which were not observed in earlier repository isolates but had been observed in other countries. The NS2 protein associates with cellular proteins that are involved in essential processes in the nucleus such as chromatin modification and the DNA damage response and therefore has a potentially important role in chromatin remodeling and other interactions required for viral replication [61].
In agreement with the other studies [35, 62], no evidence of genetic recombination within the VP2 gene was found in any of the CPV isolates analyzed. While recombination can play an essential role in the evolution of many viruses, the absence of any recombination events in CPV strains suggests that point mutations and selection pressure might be more influential in shaping the genetic diversity of CPV and the emergence of variants.
Selective pressure analysis indicated that, although the recent CPV strains were generally under negative (purifying) selection, individual potential positive selection sites could be identified in the VP1-VP2 and NS1 genes. A statistically significant increase in the dN/dS ratio was detected over the years, as was reported previously by Nguyen et al. [63]. Fu et al. [35] identified 17 positive selection sites in the VP2 gene of CPV. The NS1 gene has more positively selected sites than VP2, and specific amino acids in the NS1 gene have been associated with the differentiation of FPV and CPV-2 [51]. Positive selection at several sites in the recent CPV strains might have contributed to the fitness or adaptability of the virus in the current environment.
B-cell epitope analysis of the VP2 protein predicted that, in addition to the seven amino acid variations observed in our repository isolates (2009–2019), the majority of the recent CPV isolates (2023) had two additional variations in the major peptide epitope at amino acid residues 370 and 426 when compared to the vaccine strain. Amino acid 426 is located near the 3-fold spike of the CPV-2 capsid protein and might therefore affect the major antigenic region and result in the selection of new variants of CPV-2, posing a potential threat to domestic dogs. We speculate that the mutation at residue 426, within the major antigenic region of CPV-2, provides the mutant with certain evolutionary advantages, such as immune escape or further adaptation to the canine host [5, 64]. Hao et al. [36] predicted that mutation of residue 370, which is located at the edge of the protein-binding pocket of VP2, could significantly impact specific protein interactions affecting the binding ability of VP2. Therefore, these variations observed in the recent CPV strains at the major epitope sites could be responsible for antigenic variations and possible mismatching between the circulating CPV strains and the current CPV vaccine strain available in India.
Conclusion
In this pan-Indian study, we analyzed CPV strains collected from all geographical regions of India in 2023 and found that the CPV-2c strain was the predominant strain circulating in India, with a few co-circulating CPV-2a variants. The CPV-2b strain has almost become extinct. The study also revealed that the CPV-2c strain had almost completely replaced the previously predominant CPV-2a variant in India. The results also showed that the Q370R variation was consistently present in nearly all of the recent CPV-2c isolates (CPV-2c 370Arg variant) and some recent CPV-2a variants. Genomic analysis also confirmed that the prevailing CPV-2c strain in India probably originated in the neighboring Asian countries that border the North-Eastern region of India. Comparison of whole genome sequences with those of the repository CPV isolates (2009–2019 isolates) and the current Indian CPV vaccine strain revealed new substitutions and identified several new positive selection sites in the VP1 (N-terminus), VP2, NS1, and NS2 genes of recent CPV strains. These findings suggest the emergence of new CPV-2c variants with varied antigenic and replication properties. Although most of the sites within the VP1-VP2 and NS1 genes of the circulating CPV strains (2023) are negatively selected, an increasing dN/dS ratio over the years indicates increasing positive selective pressure. Phylogenetic analysis revealed the existence of three clades of CPV-2c strains. The predominant CPV-2c 370Arg variants were grouped with the Chinese and Nigerian CPV-2c strains and separated from the CPV vaccine strain and repository isolates. Nine amino acid variations, including two new ones, were identified in the VP2 protein. These variations are present in the four potential linear B-cell epitope regions of the predominantly circulating CPV-2c 370Arg variants. These variations may be responsible for the frequent vaccine failures observed in the field in recent times. Therefore, this pan-Indian study covering diverse geo-climatic regions of India lays the foundation for further research concerning the dynamics of virus evolution and the effects of genetic mutations. Further studies on molecular evolutionary relationships of CPV strains may help us to control potential future epidemics associated with this virus more efficiently and also aid in the development of new candidate vaccine strains.
References
Dowgier G, Lahoreau J, Lanave G, Losurdo M, Varello K, Lucente MS, Ventriglia G, Bozzetta E, Martella V, Buonavoglia C, Decaro N (2018) Sequential circulation of canine adenoviruses 1 and 2 in captive wild carnivores, France. Vet Microbiol 221:67–73
Touihri L, Bouzid I, Daoud R, Desario C, Goulli AFE, Decaro N, Ghorbel A, Buonavoglia C, Bahloul C (2009) Molecular characterization of canine parvovirus-2 variants circulating in Tunisia. Virus Genes 38:249–258
Thomson GW, Gagnon AN (1978) Canine gastroenteritis associated with a parvovirus-like agent. Can Vet J 19:346
Mira F, Purpari G, Lorusso E, Bella SD, Gucciardi F, Desario C, Macaluso G, Decaro N, Guercio A (2018) Introduction of Asian canine parvovirus in Europe through dog importation. Transbound Emerg Dis 65:16–21
Zhou P, Zeng W, Zhang X, Li S (2017) The genetic evolution of canine parvovirus–A new perspective. PLoS ONE 12:e0175035
Kelly WR (1978) An enteric disease of dogs resembling feline panleucopaenia. Aust Vet J 54:593
Parrish CR, Aquadro CF, Carmichael LE (1988) Canine host range and a specific epitope map along with variant sequences in the capsid protein gene of canine parvovirus and related feline, mink, and raccoon parvoviruses. Virol 166:293–307
Martella V, Decaro N, Elia G, Buonavoglia C (2005) Surveillance activity for canine parvovirus in Italy. J Vet Med B Infect Dis Vet Public Health 52:312–315
Buonavoglia C, Martella V, Pratelli A, Tempesta M, Cavalli A, Buonavoglia D, Bozzo G, Elia G, Decaro N, Carmichael L (2001) Evidence for evolution of canine parvovirus type 2 in Italy. J Gen Virol 82:3021–3025
Organtini LJ, Allison AB, Lukk T, Parrish CR, Hafenstein S (2015) Global displacement of canine parvovirus by a host-adapted variant: structural comparison between pandemic viruses with distinct host ranges. J Virol 89:1909–1912
Srinivas VMV, Mukhopadhyay HK, Thanislass J, Antony PX, Pillai RM (2013) Molecular epidemiology of canine parvovirus in southern India. Vet World 6:744–749
Mukhopadhyay HK, Matta SL, Amsaveni S, Antony PX, Thanislass J, Pillai RM (2014) Phylogenetic analysis of canine parvovirus partial VP2 gene in India. Virus Genes 48:89–95
Han SC, Guo HC, Sun SQ, Shu L, Wei YQ, Sun DH, Cao SZ, Peng GN, Liu XT (2015) Full-length genomic characterizations of two canine parvoviruses prevalent in Northwest China. Arch Microbiol 197:621–626
Khadse MB, Rambhau S, Kolangath S (2023) Molecular detection and phylogenetic analysis of canine parvovirus-2 in dogs. Ind J Vet Sci and Biotech 19:54–57
Parrish CR, Aquadro CF, Strassheim ML, Evermann JF, Sgro JY, Mohammed HO (1991) Rapid antigenic type replacement and DNA sequence evolution of canine parvovirus. J Virol 65:6544–6552
Raj JM, Mukhopadhyay HK, Thanislass J, Antony PX, Pillai RM (2010) Isolation, molecular characterization and phylogenetic analysis of canine parvovirus. Infect Genet Evol 10:1237–1241
Mittal M, Chakravarti S, Mohapatra JK, Chug PK, Dubey R, Upmanuyu V, Narwal PS, Kumar A, Churamani CP, Kanwar NS (2014) Molecular typing of canine parvovirus strains circulating from 2008 to 2012 in an organized kennel in India reveals the possibility of vaccination failure. Infect Genet Evol 23:1–6
Nookala M, Mukhopadhyay HK, Sivaprakasam A, Balasubramanian B, Antony PX, Thanislass J, Srinivas MV, Pillai RM (2016) Full-length VP2 gene analysis of canine parvovirus reveals emergence of newer variants in India. Acta Microbiol Immunol Hung 63:411–426
Vasu J, Srinivas MV, Antony PX, Thanislass J, Padmanaban V, Mukhopadhyay HK (2019) Comparative immune responses of pups following modified live virus vaccinations against canine parvovirus. Vet World 12:1422–1427
Karanam B, Srinivas MV, Jayalakshmi V, Antony PX, Karuppiah R, Shanmugam VP, Mukhopadhyay HK (2022) Phylodynamic and genetic diversity of parvoviruses of cats in southern India. Virus Dis 33:108–113
Hao X, He Y, Wang C, Xiao W, Liu R, Xiao X, Zhou P, Li S (2020) The increasing prevalence of CPV-2c in domestic dogs in China. Peer J 29(8):e9869
Hu W, Xu X, Liu Q, Ji J, Kan Y, Yao L, Bi Y, Xie Q (2020) Molecular characterisation and genetic diversity of canine parvovirus type 2 prevalent in Central China. J Vet Res 64:347–354
Jiang H, Yu Y, Yang R, Zhang S, Wang D, Jiang Y, Yang W, Huang H, Shi C, Ye L, Yang G, Wang J, Wang C (2021) Detection and molecular epidemiology of canine parvovirus type 2 (CPV-2) circulating in Jilin Province, Northeast China. Comp Immunol Microbiol Infect Dis 74:101602
Tamura K, Stecher G, Kumar S (2021) MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Ohshima T, Hisaka M, Kawakami K, Kishi M, Tohya Y, Mochizuki M (2008) Chronological analysis of canine parvovirus type 2 isolates in Japan. J Vet Med Sci 70:769–775
Yi L, Tong M, Cheng Y, Song W, Cheng S (2016) Phylogenetic analysis of canine parvovirus VP2 Gene in China. Transbound Emerg Dis 63:e262–e269
Geng Y, Guo D, Li C, Wang E, Wei S, Wang Z, Yao S, Zhao X, Su M, Wang X, Wang J, Wu R, Feng L, Sun D (2015) Co-circulation of the rare CPV-2c with unique Gln370Arg substitution, new CPV-2b with unique Thr440Ala substitution, and new CPV-2a with high prevalence and variation in Heilongjiang Province. Northeast China. PloS One 10:e0137288
Zhou L, Tang Q, Shi L, Kong M, Liang L, Mao Q, Bu B, Yao L, Zhao K, Cui S, Leal E (2016) Full-length genomic characterization and molecular evolution of canine parvovirus in China. Virus Genes 52:411–416
Kumari GD, Pushpa RR, Subramanyam KV, Rao TS, Satheesh K, Srinivas VMV (2021) Molecular characterization and phylogenetic analysis of canine parvovirus strains in Andhra Pradesh, India. Indian J Anim Health 60:90–97
Chinchkar SR, Subramanian BM, Rao NH, Rangarajan PN, Thiagarajan D, Srinivasan VA (2006) Analysis of VP2 gene sequences of canine parvovirus isolates in India. Arch Virology 151:1881–1887
Kulkarni MB, Deshpande AR, Gaikwad SS, Majee SB, Suryawanshi PR, Awandkar SP (2019) Molecular epidemiology of canine parvovirus shows CPV-2a genotype circulating in dogs from western India. Infect Genet Evol 75:103987
Singh M, Tripathi P, Singh S, Sachan M, Chander V, Sharma GK, De UK, Kota S, Putty K, Singh RK, Nara S (2021) Identification and characterization of DNA aptamers specific to VP2 protein of canine parvovirus. Appl Microbiol Biotechnol 105:8895–8906
Magar SA, Warke SR, Tumlam UM, Bhojne GR, Ingle VC (2020) Molecular typing of canine parvovirus-2 occurring in Nagpur by multiplex PCR. J Entomol Zool Stud 8:91–95
Abhiram S, Mondal T, Samanta S, Batabyal K, Joardar SN, Samanta I, Isore DP, Dey S (2023) Occurrence of canine parvovirus type 2c in diarrhoeic faeces of dogs in Kolkata, India. Virus Dis 34:339–344
Fu P, He D, Cheng X, Niu X, Wang C, Fu Y, Li K, Zhu H, Lu W, Wang J, Chu B (2022) Prevalence and characteristics of canine parvovirus type 2 in Henan Province, China. Microbiol Spectr 10:e01856-e1922
Hao X, Li Y, Xiao X, Chen B, Zhou P, Li S (2022) The changes in canine parvovirus variants over the years. Int J Mol Sci 23:11540
Guo L, Yang SL, Chen SJ, Zhang Z, Wang C, Hou R, Ren Y, Wen X, Cao S, Guo W, Hao Z, Quan Z, Zhang M, Yan QG (2013) Identification of canine parvovirus with the Q370R point mutation in the VP2 gene from a giant panda (Ailuropoda melanoleuca). Virol J 10:1–7
Vannamahaxay S, Vongkhamchanh S, Intanon M, Tangtrongsup S, Tiwananthagorn S, Pringproa K, Chuammitri P (2017) Molecular characterization of canine parvovirus in Vientiane, Laos. Arch Virol 162:1355–1361
Lin YC, Chiang SY, Wu HY, Lin JH, Chiou MT, Liu HF, Lin CN (2017) Phylodynamic and genetic diversity of canine parvovirus type 2c in Taiwan. Int J Mol Sci 18:2703
Moon BY, Jang J, Kim SH, Kim YH, Lee HK, So BJ, Park CK, Lee KK (2020) Genetic characterization of canine parvovirus type 2c from domestic dogs in Korea. Transbound Emerg Dis 67:1645–1653
Chen B, Zhang X, Zhu J, Liao L, Bao E (2021) Molecular epidemiological survey of canine parvovirus circulating in China from 2014 to 2019. Pathog 10:588
Wang J, Lin P, Zhao H, Cheng Y, Jiang Z, Zhu H, Wu H, Cheng S (2016) Continuing evolution of canine parvovirus in China: Isolation of novel variants with an Ala5Gly mutation in the VP2 protein. Infect Genet Evol 38:73–78
Chiang SY, Wu HY, Chiou MT, Chang MC, Lin CN (2016) Identification of a novel canine parvovirus type 2c in Taiwan. Virol J 13:1–7
Hoang M, Lin W, Le VP, Nga BTT, Chiou MT, Lin CN (2019) Molecular epidemiology of canine parvovirus type 2 in Vietnam from November 2016 to February 2018. Virol J 16:52
Jantafong T, Ruenphet S, Garner HR, Ritthipichai K (2022) Tracing the genetic evolution of canine parvovirus type 2 (CPV-2) in Thailand. Pathog 11:1460
Chapman MS, Rossmann MG (1993) Structure, sequence, and function correlations among parvoviruses. Virol 194:491–508
Jeoung SY, Ahn SJ, Kim D (2008) Genetic analysis of VP2 gene of canine parvovirus isolates in Korea. J Vet Med Sci 70:719–722
Phromnoi S, Sirinarumitr K, Sirinarumitr T (2010) Sequence analysis of VP2 gene of canine parvovirus isolates in Thailand. Virus Genes 41:23–29
Maya L, Calleros L, Francia L, Hernández M, Iraola G, Panzera Y, Sosa K, Pérez R (2013) Phylodynamics analysis of canine parvovirus in Uruguay: evidence of two successive invasions by different variants. Arch Virology 158:1133–1141
Soma T, Taharaguchi S, Ohinata T, Ishii H, Hara M (2013) Analysis of the VP2 protein gene of canine parvovirus strains from affectd dogs in Japan. Res Vet Sci 94:368–371
Mira F, Purpari G, Bella SD, Colaianni ML, Schirò G, Chiaramonte G, Gucciardi F, Pisano P, Lastra A, Decaro N, Guercio A (2019) Spreading of canine parvovirus type 2c mutants of Asian origin in southern Italy. Transbound Emerg Dis 66:2297–2304
Alexis VA, Sonia V, Daniela S, Miguel G, Timothy H, Valentina F, Lisette L, Leonardo S (2021) Molecular analysis of full-length VP2 of canine parvovirus reveals antigenic drift in CPV-2b and CPV-2c variants in Central Chile. Animals (Basel) 11:2387
Abayli H, Aslan O, Tumer KC, Can-Sahna K, Tonbak S (2022) Predominance and first complete genomic characterization of canine parvovirus 2b in Turkey. Arch Virol 167:1831–1840
Li S, Chen X, Hao Y, Zhang G, Lyu Y, Wang J, Liu W, Qin T (2022) Characterization of the VP2 and NS1 genes from canine parvovirus type 2(CPV-2) and feline panleukopenia virus (FPV) in Northern China. Front Vet Sci 9:934849
Li G, Ji S, Zhai X, Zhang Y, Liu J, Zhu M, Zhou J, Su S (2017) Evolutionary and genetic analysis of the VP2 gene of canine parvovirus. BMC Genom 18:534
Zhuang QY, Qiu Y, Pan ZH, Wang SC, Wang B, Wu WK, Yu JM, Yi Y, Sun FL, Wang KC (2019) Genome sequence characterization of canine parvoviruses prevalent in the Sichuan province of China. Transbound Emerg Dis 66:897–907
Zádori Z, Szelei J, Lacoste MC, Li Y, Gariépy S, Raymond P, Allaire M, Nabi IR, Tijssen P (2001) A viral phospholipase A2 is required for parvovirus infectivity. Dev cell 1:291–302
Vihinen-Ranta M, Wang D, Weichert WS, Parrish CR (2002) The VP1 N-terminal sequence of canine parvovirus affects nuclear transport of capsids and efficient cell infection. J Virol 76:1884–1891
Ogbu KI, Mira F, Purpari G, Nwosuh C, Loria GR, Schirò G, Chiaramonte G, Tion MT, Bella SD, Ventriglia G, Decaro N, Anene BM, Guercio A (2020) Nearly full-length genome characterization of canine parvovirus strains circulating in Nigeria. Transbound Emerg Dis 67:635–647
Temizkan MC, Temizkan S (2023) Canine parvovirus in Turkey: First whole-genome sequences, strain distribution, and prevalence. Viruses 15:957
Mattola S, Salokas K, Aho V, Mäntylä E, Salminen S, Hakanen S, Niskanen EA, Svirskaite J, Ihalainen TO, Airenne KJ, Kaikkonen-Määttä M, Parrish CR, Varjosalo M, Vihinen-Ranta M (2022) Parvovirus non structural protein 2 interacts with chromatin-regulating cellular proteins. PLoS Pathog 18:e1010353
Silva SP, Silva L, Rodrigues E, Cardoso JF, Tavares FN, Souza WM, Santos CMP, Martins FMS, Jesus IS, Brito TC, Moura TPC, Nunes MRT, Casseb LMN, Filho ES, Casseb AR (2017) Full-length genomic and molecular characterization of canine parvovirus in dogs from North of Brazil. Genet Mol Res 16:gmr16039719
Manh TN, Piewbang C, Rungsipipat A, Techangamsuwan S (2021) Molecular and phylogenetic analysis of Vietnamese canine parvovirus 2C originated from dogs reveals a new Asia-IV clade. Transbound Emerg Dis 68:1445–1453
Allison AB, Organtini LJ, Zhang S, Hafenstein SL, Holmes EC, Parrish CR (2016) Single mutations in the VP2 300 loop region of the three-fold spike of the carnivore parvovirus capsid can determine host range. J Virol 90:753–767
Acknowledgments
We thank the Dean, Rajiv Gandhi Institute of Veterinary Education and Research (RIVER), Puducherry, India, for providing the necessary funds and facilities to carry out this study.
Funding
Funding and facilities for this research were received from the Rajiv Gandhi Institute of Veterinary Education & Research, Puducherry–605 009, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The work described in this article involved the use of non-experimental (owned or unowned) animals. Approval from the Institutional Animal Ethics Committee (IAEC) to carry out this study was not required, as no invasive techniques were used. Faecal samples were collected from CPV/FPV-suspected or apparently healthy animals from veterinary hospitals, adoption centers, and animal rescue shelters in India. Established internationally recognized high standards (‘best practice’) of veterinary clinical care for the individual patients were consistently followed. Ethical approval from a committee was therefore not explicitly required.
Informed consent
Informed verbal consent was obtained from the owner or legal custodian of all animals described in this work (non-experimental animals) to collect samples for testing for parvovirus diagnosis.
Conflict of interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Additional information
Handling Editor: Ana Cristina Bratanich
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Reddy, H., Srinivas, M.V., Vasu, J. et al. Whole-genome sequence analysis of canine parvovirus reveals replacement with a novel CPV-2c strain throughout India. Arch Virol 169, 189 (2024). https://doi.org/10.1007/s00705-024-06096-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-024-06096-2