
Abstract
Genotype 4 (G4) Eurasian avian-like lineage swine H1N1 influenza A viruses, which are reassortants containing sequences from the pandemic 2009 H1N1 virus lineage, triple-reassortant-lineage internal genes, and EA-lineage external genes, have been reported in China since 2013. These have been predominant in pig populations since 2016 and have exhibited pandemic potential. In this study, we developed a one-step multiplex RT-qPCR assay targeting the M, HA1, and PB2 genes to detect G4 and related EA H1N1 viruses, with detection limits of 1.5 × 101 copies/μL and 1.15 × 10−2 ng/μL for the purified PCR products and RNA templates, respectively. The specificity of the detection method was confirmed using various influenza virus subtypes. When the one-step multiplex RT-qPCR assay was applied to swine respiratory samples collected between 2020 and 2022 in Korea, a virus related to G4 EA H1N1 strains was detected. Phylogenetic analysis based on portions of all eight genome segments showed that the positive sample contained HA, NA, PB2, NS, and NP genes closely related to those of G4 EA H1N1 viruses, confirming the ability of our assay to accurately detect G4 EA H1N1 viruses in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Influenza A virus (IAV) is a pathogen that can cause severe illnesses in mammals and birds [10]. Because the RNA genome of IAV is segmented, gene reassortment occurs frequently, resulting in the emergence of novel viruses [23]. Most of the IAVs in past pandemics were reassortants resulting from the shuffling of genes between human and animal IAVs [2, 11]. Among the mammals that can transmit influenza viruses, pigs are considered “mixing vessels” that might contribute to the emergence of novel influenza viruses with pandemic potential, as swine can be infected by both human- and avian-origin influenza viruses [16, 26]. Notable examples include the “Spanish” flu pandemic of 1918 and the H1N1 (pdm/09 H1N1) pandemic of 2009 [4, 27]. Thus, constant surveillance of IAVs in pig populations should be conducted to prepare for future pandemic IAV threats.
Eurasian avian-like (EA) swine H1N1 viruses are derived from an avian influenza virus [21] and have caused diseases endemic to Europe and Asia [5, 13, 20, 28]. Recently, researchers demonstrated that the genotype 4 (G4) EA H1N1 virus, predominant since 2016 in pig populations in China, has the potential to cause an influenza pandemic [24]. The G4 EA H1N1 virus contains sequences from the pdm/09 H1N1 lineage, triple reassortant (TR)-lineage internal genes, and EA-lineage external genes [24]. G4 EA H1N1 viruses can bind to receptors on human cells and effectively replicate in human airway epithelial cells, and they have shown significant infectivity and aerosol transmission in animal models [24, 25]. In addition, G4 EA H1N1 viruses have a high risk of spreading to humans because of their low cross-reactivity with trivalent vaccines [24]. Currently, although a TaqMan-probe-based G4 EA H1N1 virus detection kit is available, it is difficult to perform IAV screening using a single tube, as the detection wavelengths of all of the probes are identical. To accurately identify the genotype of the virus, it is important to detect viruses related to G4 EA H1N1 viruses from clinical samples from swine. In addition, despite their pandemic potential, epidemiological studies of G4 EA H1N1 viruses have not been conducted in Eurasian countries other than China. Therefore, we have developed a platform for specific detection of G4 EA H1N1-related viruses in a single tube.
Materials and methods
Primers and probes
To detect G4 EA swine H1N1 viruses, we downloaded 171 HA1 gene sequences of EA-lineage swine H1N1 viruses, 29 PB2 gene sequences of G4 EA swine H1N1 viruses, and 41 PB2 gene sequences of EA swine H1N1 viruses from the GenBank and Global Initiative on Sharing Avian Influenza Data (GISAID) databases [24]. We performed ClustalW multiple alignments using BioEdit software (7.2.5 version) to target the common sequences of the EA lineage HA1 and PB2 genes of G4 EA swine H1N1 viruses. To determine the influenza virus type, we used universal primers and a probe targeting the matrix gene recommended by WHO (Supplementary Table S2).
Generation of recombinant G4 EA H1N1 virus
G4 EA H1N1 virus was rescued using a reverse genetics system kindly provided by Dr. Richard J. Webby (St. Jude Children’s Research Hospital, TN, USA). Eight segments of the A/swine/Shandong/1207/2016 (SW/SD/1207/16) virus [24] were cloned into the pHW2000 vector, and the constructs were used to transfect co-cultured human embryonic kidney (293T, CRL-3216) and Madin–Darby canine kidney (MDCK, CRL-2936) cells [8]. The culture supernatant was collected and used to inoculate 9- to 11-day-old specific-pathogen-free (SPF) chicken eggs. After virus rescue, the complete genome sequences of the rescued viruses were verified by sequencing, and all rescued viruses were stored at -80 °C until use. All viral experiments were conducted at the Korea Research Institute of Bioscience and Biotechnology (KRIBB, South Korea) and approved by and conducted in accordance with the guidelines of the Institutional Biosafety Committee (IBC, approval number KRIBB-IBC-20200213) of the KRIBB.
RNA extraction and cDNA synthesis
Recombinant SW/SD/1207/16 RNA was extracted using TRIzol LS Reagent (Invitrogen, USA), following the manufacturer’s instructions. RNA was reverse transcribed into cDNA using M-MLV reverse transcriptase (Promega, USA) with random hexamers, following the manufacturer’s protocol.
Preparation of purified PCR products for analytical sensitivity determination
To generate a quantitative standard, M, HA1, and PB2 sequences of recombinant SW/SD/1207/16 containing primer and probe binding sites were amplified by conventional PCR using the primers listed in Supplementary Table S2. M, HA1, and PB2 amplicons were separated by electrophoresis in an agarose gel and purified using an Expin Combo GP kit (GeneAll, South Korea). Each purified PCR product was quantified using a spectrophotometer (DeNovix, USA). The quantities were converted into copy numbers using the following formula [9]:
Then, the purified PCR products were serially diluted tenfold with nuclease-free water from 1.5 × 108 copies/μL to 1.5 × 101 copies/μL.
One-step multiplex RT-qPCR
All RT-qPCR reactions were performed using a SensiFAST Probe No-ROX One-Step Kit (Bioline, USA) in a volume of 20 μL. The mixture contained 4 μL of RNA template, 0.8 μL of each F/R primer (10 pmol), 0.2 μL of each probe (10 pmol), 10 μL of 2X SensiFAST Probe No-ROX One-Step Mix, 0.2 μL of reverse transcriptase, and 0.4 μL of RiboSafe RNase Inhibitor.
Multiplex RT-qPCR was performed with reverse transcription at 45 °C for 20 min and 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s and 54 °C for 30 s, using a LightCycler 96 System (Roche, USA). Diethylpyrocarbonate (DEPC)-treated buffer was used as a no-template control (NTC). The result was considered positive if the amplification curve showed a positive signal within 40 cycles.
Analytical sensitivity of one-step multiplex RT-qPCR
To evaluate the limit of detection of the newly developed multiplex RT-qPCR, we used purified PCR products and RNA templates of recombinant SW/SD/1207/16. The extracted RNA was also quantified spectrophotometrically and serially diluted tenfold from 1.15 ng/μL to 1.15 × 10−3 ng/μL. We also estimated the sensitivity of the G4 EA H1N1 Swine Flu Virus RT-qPCR Detection Kit (ScienCell, USA), following the manufacturer’s manual, for comparison with the sensitivity of our newly developed multiplex RT-qPCR.
Analytical specificity of one-step multiplex RT-qPCR
The analytical specificity was determined for different IAV subtypes and other viruses. We used nucleic acid samples extracted from human H1N1 virus, Eurasian avian lineage H1N1 virus, other IAV subtypes (H3N2, H3N8, and H9N2 viruses), porcine reproductive and respiratory syndrome virus (PRRSV), human coronavirus OC43 (HCoV-OC43) (Korea Bank for Pathogenic Virus, Seoul, South Korea), porcine epidemic diarrhoea virus (PEDV), porcine circovirus type 3 (PCV3), and mammalian orthoreovirus (MRV) to assess the specificity of the newly developed one-step multiplex RT-qPCR. The human H1N1 virus and other subtypes were provided by the College of Veterinary Medicine, Chonnam National University.
Clinical samples from swine
We performed IAV screening on 33 IAV-positive and 30 IAV-negative archived swine respiratory samples provided by OptiPharm Inc. (Osong, South Korea), using universal primers and probe sets specific for the IAV matrix gene, followed by screening using our multiplex RT-qPCR. We then compared the results of the first and second screenings.
Partial sequencing and phylogenetic analysis
We performed conventional PCR using universal HA1 and PB2 primers (Supplementary Table S3) for partial sequencing and genetic lineage determination of viruses in swine clinical samples. Sanger sequencing was performed using an Applied Biosystems 3730xl DNA Analyzer. Partial HA1 sequences were analyzed using the Swine H1 Clade Classification Tool in the Influenza Research Database. To examine the genetic lineage in detail, partial PB2 and HA sequences were analyzed using BLASTn (https://blast.ncbi.nlm.nih.gov/) and Molecular Evolutionary Genetics Analysis 11 (MEGA 11). Phylogenetic trees based on these nucleotide sequences were constructed using the maximum-likelihood method with 1000 replicates in MEGA 11. We also performed partial sequencing of the NA1 and M genes of the Cy5-signal-positive samples. Reference sequences from the GenBank and GISAID databases were included in the phylogenetic trees.
Further sequence analysis of G4 EA H1N1 virus-positive sample
Samples that yielded a positive fluorescence signal were further analyzed by partial sequencing of the PB1, PA, NP, and NS genes, using the primers listed in Supplementary Table S3.
Virus isolation
MDCK cells were grown in minimum essential medium (MEM) with 5% fetal bovine serum (FBS) and 1X antibiotic-antimycotic in 25-cm2 flasks. For virus isolation, samples that showed a Cy5 fluorescent signal were passed through a 0.45-μm syringe filter, diluted tenfold, and applied to a cell monolayer for 1 h at 37 °C in 5% CO2. The cells were then washed once with DPBS, after which MEM with 0.3 μg of TPCK-trypsin per mL was added to the cell monolayer, and the cells were incubated for 96 h. After incubation, the cell supernatants were passaged, and the presence of IAV was confirmed using RT-qPCR.
Results
Analytical sensitivity of one-step multiplex RT-qPCR
We estimated the sensitivity of multiplex RT-qPCR using purified PCR products and RNA templates, and the experiment was repeated at least three times. For the diluted purified PCR products, the detection limit of RT-qPCR was 1.5 × 101 copies/μL based on the amplification curves produced under singleplex and multiplex conditions (Supplementary Fig. S1) and the electrophoresis results of singleplex RT-qPCR (Supplementary Fig. S2). The standard curves produced by RT-qPCR showed linearity (R2 = 0.9903–0.999, Supplementary Fig. S1). In the case of diluted RNA, the detection limit of one-step multiplex RT-qPCR was 1.15 × 10−1 ng/μL (Table 2). The NTC did not produce amplification curves. When the multiplex RT-qPCR method was compared with the detection kit, our multiplex RT-qPCR was found to be tenfold more sensitive. Considering these results, we considered a result positive for the M and HA1 genes when the Cq value was less than 36 and positive for the PB2 gene when the Cq value was less than 35.
Analytical specificity of one-step multiplex RT-qPCR
The specificity of the multiplex RT-qPCR was assessed in triplicate using different IAV subtypes and other pathogens. We found that all IAV subtypes were detected by the primer/probe set targeting the matrix gene, while other subtypes and pdm/09 H1N1 lineage human H1N1 viruses were not detected by our HA1 and PB2 primers and probe sets (Table 3). Fluorescent signals were not detected for PRRSV, HCoV-OC43, PEDV, PCV-3, or MRV.
Application of one-step multiplex RT-qPCR using swine clinical samples
Thirty-three IAV-positive and 30 IAV-negative clinical swine samples provided by OptiPharm Inc. were used to screen for swine IAVs. Positive and negative samples were verified using universal matrix gene primers and probe sets (Supplementary Table S4). The newly developed one-step multiplex RT-qPCR showed that all of the positive samples were IAV-positive, and two of the 30 negative samples showed fluorescent signals but did not show the expected DNA band on the agarose gel. Among the positive samples, seven were HEX signal-positive, six were Cy5 signal-positive, and one (named ‘6IP’) showed all of the signals (Supplementary Table S4). We regarded 6IP as infected with the G4 EA H1N1 virus.
Partial sequencing and phylogenetic analysis
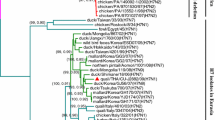
Partial sequences of swine influenza viruses were deposited in the GenBank database (Supplementary Table S1). We performed conventional PCR using the primers listed in Supplementary Table S3 for partial sequencing and examined the genetic lineage of the partial HA1 and PB2 genes using the MEGA 11 program for all swine clinical samples. We further sequenced the positive samples and analyzed the sequences using the BLASTn and MEGA 11 programs (Supplementary Table S4 and Supplementary Fig. S5). We also analyzed partial HA1 and PB2 sequences from 13 positive samples that showed fluorescent signals. Among the eight HEX-signal-positive samples, five were assigned to the 1C.2.3 clade by the Influenza Research Database (Fig. 1). Among the six Cy5-signal-positive samples, five belonged to the pdm/09 H1N1 lineage. When partial NA sequences were analyzed, three samples belonged to the pdm/09 H1N1 lineage, and one sample belonged to the EA lineage. When partial M sequences were analyzed, four samples belonged to the pdm/09 H1N1 lineage, and one sample belonged to the EA lineage (Supplementary Table S4 and Supplementary Fig. S3).

HA1 clade assignment of swine clinical samples using the Influenza Research Database (https://www.fludb.org)
Further phylogenetic analysis of sample 6IP showed that the PB1 and NP genes of 6IP belonged to the pdm/09 H1N1 lineage, whereas the PA and NS genes belonged to the avian and TR lineage, respectively. The partial PB2, HA, NP, and NS sequences appear to be related to those of G4 EA H1N1 viruses (Supplementary Figs. S3 and S4). We attempted to isolate the G4 EA H1N1 virus-related swine influenza viruses using MDCK cells, but we were unsuccessful.
Discussion
The goal of this study was to efficiently detect G4 and related EA H1N1 viruses and confirm the circulation of G4 EA H1N1 viruses in pigs in South Korea. EA swine H1N1 viruses have been circulating in pig populations on the Eurasian continent [20]. Most human infections caused by H1N1 viruses are acquired by direct exposure to pigs, and EA swine H1N1 viruses occasionally cause severe human infections [5, 17, 19]. A previous study also demonstrated that EA H1N1 viruses could become more pathogenic by accumulating mutations in the polymerase gene [18]. While EA swine H1N1 viruses have been circulating in Eurasian countries, G4 EA H1N1 viruses have emerged in China and have been predominant since 2016 [25]. G4 EA H1N1 viruses have severe pathogenicity, enhanced replication, and transmission ability in vivo and can be transmitted from pigs to humans [6, 14, 24].
Despite the pandemic potential of these viruses, few G4 EA H1N1 virus detection tools exist. For genotyping of IAV, the genetic lineage of all eight segments needs to be identified. To reduce surveillance time, we needed to be able to select G4 EA H1N1 virus-positive samples from among clinical samples. qPCR based on TaqMan probes has high sensitivity and specificity and good reproducibility for molecular diagnosis in the veterinary field [3]. Thus, we developed a multiplex RT-qPCR method for surveillance of IAVs using a single tube. In a previous study, the PB2 and HA genes were shown to be major determinants of the host specificity of IAV [15]. The general RT-qPCR method targeting matrix genes is the gold standard for the rapid molecular detection and determination of influenza virus types [22]. We considered these studies to design primers and probes. M, HA1, and PB2 genes were used to identify influenza virus types, verify serotypes, and narrow the host range of IAV, respectively.
Using the primers and probes described here, it was possible to detect G4 EA H1N1 viruses at 1.5 × 101 copies/μL when using purified PCR products as templates (Table 1 and Supplementary Fig. S1). When RNA was used as a template, the detection limit of multiplex RT-qPCR was 1.15 × 10−1 ng/μL (Table 2). Under the experimental conditions used in this study, the newly developed assay was approximately tenfold more sensitive than the commercial RT-qPCR kit. Based on previous results showing that the detection limit of triplex RT-qPCR for the detection of avian IAVs was 5.0 × 101 copies/μL [12], our multiplex RT-qPCR is suitable for veterinary use, with adequate analytical sensitivity.
In terms of analytical specificity, the newly developed multiplex RT-qPCR detects a narrow range of influenza viruses. Although the A/California/04/2009(H1N1), A/Puerto Rico/8/1934(H1N1), and A/wild bird/Korea/SK14/2014(H1N1) viruses have identical subtypes, the multiplex RT-qPCR detects only the Eurasian avian lineage, which is closely related to the EA lineage [1, 7] or EA lineage HA1 gene (Tables 2 and 3). Interestingly, our PB2 primer and probe set used for G4 EA swine H1N1 viruses did not detect the A/California/04/2009(H1N1) virus, although the genetic lineage of the PB2 gene of the virus is the same as that of the G4 EA H1N1 virus [24] (Tables 2 and 3). This suggests that the PB2 primers and probe are highly specific for swine-origin IAVs.
In a comparison of the IAV screening results with the multiplex RT-qPCR results, the matrix gene primers and probe sets showed 100% agreement with 33 IAV-positive samples. However, two IAV-negative samples gave false-positive results (Supplementary Table S4). To verify the false positive and false negative results obtained by multiplex RT-qPCR, we identified the genetic lineages of the HA1 and PB2 genes by partial sequencing. This showed that our primers and probe set targeting the EA lineage HA1 gene produced a higher rate of false-positive results than those targeting the pdm/09 H1N1 lineage PB2 gene (Table 4). Although nonspecific reactions might have occurred due to the low annealing temperature and degenerate probe sequences, considering its relatively high analytical sensitivity, the multiplex RT-qPCR developed in this study might still be suitable for surveillance of G4 EA H1N1 and related viruses.
We selected a G4 EA H1N1 virus-positive sample (6IP) and performed a phylogenetic analysis. The partial PB2, HA, NP, NA, and NS sequences of 6IP appeared to be related to those of G4 EA H1N1 viruses. This sample was collected on January 27, 2022, in South Korea, and the results might therefore imply a reassortment of G4 EA H1N1 viruses circulating in swine populations in Asia. A gene reassortment event with the G4 EA H1N1 virus genome was reported in China in 2020 [29]. G4 EA H1N1 viruses have mainly been reported in China since 2016. However, in countries other than China, no cases of detection and isolation of G4 EA H1N1 viruses from swine populations have been reported. Phylogenetic analysis of the 6IP sample showed evidence of the circulation of G4 EA H1N1 viruses in swine populations in South Korea. However, because our attempts to isolate the G4 EA H1N1 virus-related swine IAV were unsuccessful, we could not obtain a whole-genome sequence of the virus in the 6IP sample for a more precise analysis of the reassortment of G4 EA H1N1 viruses. Therefore, there should be additional follow-up studies using embryonated egg inoculation to isolate and characterize G4 EA H1N1 viruses in Korea.
In conclusion, we developed and validated a one-step multiplex RT-qPCR assay that can potentially be used to screen for G4 EA swine influenza viruses. Applying the one-step multiplex RT-qPCR assay for viral screening of swine respiratory samples collected in Korea between 2020 and 2022, we identified one sample (6IP) that was positive for a G4 EA H1N1-related virus. Although we failed to isolate the virus, phylogenetic analysis based on partial sequences of all eight gene segments showed that the positive sample 6IP contained nucleic acids of IAV HA, NA, PB2, NS, and NP genes that were closely related to those of G4 EA H1N1 viruses. This suggests that G4 EA H1N1 viruses or related reassortant viruses might be circulating in the swine population of Korea. Therefore, additional screening for G4 EA H1N1 and related viruses should be performed in future epidemiological studies.
Data availability
The sequences generated in this study were deposited in the GenBank database under the accession numbers listed in Supplementary Table S1.
References
Anderson TK, Macken CA, Lewis NS et al (2016) A phylogeny-based global nomenclature system and automated annotation tool for H1 hemagglutinin genes from swine influenza A viruses. mSphere 1(6):e00275-00216
Belshe RB (2005) The origins of pandemic influenza–lessons from the 1918 virus. N Engl J Med 353(21):2209–2211
Chang CY, Deng MC, Wang FI et al (2014) The application of a duplex reverse transcription real-time PCR for the surveillance of porcine reproductive and respiratory syndrome virus and porcine circovirus type 2. J Virol Methods 201:13–19
Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, Dawood FS, Jain S et al (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360(25):2605–2615
Freidl GS, Meijer A, de Bruin E et al (2014) Influenza at the animal-human interface: a review of the literature for virological evidence of human infection with swine or avian influenza viruses other than A(H5N1). Euro Surveill 19(18):1
Gu M, Chen K, Ge Z et al (2022) Zoonotic threat of G4 genotype Eurasian avian-like swine influenza A(H1N1) Viruses, China, 2020. Emerg Infect Dis 28(8):1664–1668
Guo F, Yang J, Pan J et al (2020) Origin and evolution of H1N1/pdm2009: A codon usage perspective. Front Microbiol 11:1615
Hoffmann E, Neumann G, Kawaoka Y et al (2000) A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci USA 97(11):6108–6113
Huang YL, Pang VF, Pan CH et al (2009) Development of a reverse transcription multiplex real-time PCR for the detection and genotyping of classical swine fever virus. J Virol Methods 160(1–2):111–118
Javanian M, Barary M, Ghebrehewet S et al (2021) A brief review of influenza virus infection. J Med Virol 93(8):4638–4646
Krammer F, Smith GJD, Fouchier RAM et al (2018) Influenza. Nat Rev Dis Primers 4(1):3
Le TB, Kim HK, Na W et al (2020) Development of a multiplex RT-qPCR for the detection of different clades of avian influenza in poultry. Viruses 12(1):1
Li X, Guo L, Liu C et al (2019) Human infection with a novel reassortant Eurasian-avian lineage swine H1N1 virus in northern China. Emerg. Microbes Infect. 8(1):1535–1545
Li Z, Zhao X, Huang W et al (2022) Etiological characteristics of the first human infection with the G4 genotype Eurasian avian⁃ like H1N1 swine influenza virus in Yunnan Province China. Chin. J. Virol. 38:290–297
Long JS, Mistry B, Haslam SM et al (2019) Host and viral determinants of influenza A virus species specificity. Nat Rev Microbiol 17(2):67–81
Ma W, Kahn RE, Richt JA (2008) The pig as a mixing vessel for influenza viruses: human and veterinary implications. J Mol Genet Med 3(1):158–166
Meng F, Chen Y, Song Z et al (2022) Continued evolution of the Eurasian avian-like H1N1 swine influenza viruses in China. Sci. China Life Sci. 1:1
Meng F, Yang H, Qu Z et al (2022) A Eurasian avian-like H1N1 swine influenza reassortant virus became pathogenic and highly transmissible due to mutations in its PA gene. Proc Natl Acad Sci USA 119(34):e2203919119
Mondiale de la Santé O (2022) World Health Organization 2022 Antigenic and genetic characteristics of zoonotic influenza A viruses and development of candidate vaccine viruses for pandemic preparedness–Caractéristiques génétiques et antigéniques des virus grippaux A zoonotiques et mise au point de virus vaccinaux candidats pour se préparer à une pandémie. Week Epidemiol Record 97:120–132
Parys A, Vandoorn E, King J et al (2021) Human infection with Eurasian avian-like swine influenza A(H1N1) Virus, the Netherlands, September 2019. Emerg Infect Dis 27(3):939–943
Pensaert M, Ottis K, Vandeputte J et al (1981) Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential importance for man. Bull World Health Organ 59(1):75–78
Spackman E, Senne DA, Myers TJ et al (2002) Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 40(9):3256–3260
Steel J, Lowen AC (2014) Influenza A virus reassortment. Curr Top Microbiol Immunol 385:377–401
Sun H, Xiao Y, Liu J et al (2020) Prevalent Eurasian avian-like H1N1 swine influenza virus with 2009 pandemic viral genes facilitating human infection. Proc Natl Acad Sci USA 117(29):17204–17210
Sun H, Liu J, Xiao Y et al (2021) Pathogenicity of novel reassortant Eurasian avian-like H1N1 influenza virus in pigs. Virology 561:28–35
Suzuki T, Horiike G, Yamazaki Y et al (1997) Swine influenza virus strains recognize sialylsugar chains containing the molecular species of sialic acid predominantly present in the swine tracheal epithelium. FEBS Lett 404(2–3):192–196
Taubenberger JK, Morens DM (2006) 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 12(1):15–22
Vijaykrishna D, Smith GJ, Pybus OG et al (2011) Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473(7348):519–522
Wang SY, Wen F, Yu LX et al (2022) Potential threats to human health from Eurasian avian-like swine influenza A(H1N1) virus and its reassortants. Emerg Infect Dis 28(7):1489–1493
Acknowledgements
This work was supported by the Bio & Medical Technology Development Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science & ICT (2021M3E5E3083402), and the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2020R1A6A1A06046235).
Funding
This article was funded by Ministry of Science and ICT, South Korea (2021M3E5E3083402), and Ministry of Education (2020R1A6A1A06046235).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Handling Editor: William G Dundon.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moon, S.H., Na, W., Shin, S. et al. Evidence for the circulation of genotype 4 Eurasian avian-like lineage swine H1N1 influenza A viruses on Korean swine farms, obtained using a newly developed one-step multiplex RT-qPCR assay. Arch Virol 168, 267 (2023). https://doi.org/10.1007/s00705-023-05887-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05887-3