
Abstract
Here, we report the characterization of a novel (−)ssRNA mycovirus isolated from Auricularia heimuer CCMJ1222, using a combination of RNA-seq, reverse transcription polymerase chain reaction, 5′ and 3′ rapid amplification of cDNA ends, and Sanger sequencing. Based on database searches, sequence alignment, and phylogenetic analysis, we designated the virus as “Auricularia heimuer negative-stranded RNA virus 1” (AhNsRV1). This virus has a monopartite RNA genome related to mymonaviruses (order Mononegavirales). The AhNsRV1 genome consists of 11,441 nucleotides and contains six open reading frames (ORFs). The largest ORF encodes a putative RNA-dependent RNA polymerase; the other ORFs encode hypothetical proteins with no conserved domains or known function. AhNsRV1 is the first (−)ssRNA virus and the third virus known to infect A. heimuer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Fungal viruses are prevalent in various fungi, including yeasts, mushrooms, and pathogenic fungi that infect plants, insects, and humans [1,2,3]. In 1948, the first fungal virus was found in the edible fungus Agaricus bisporus [4]. Examples of other fungal viruses reported in other mushroom species include mushroom bacilliform virus (MBV) and mushroom virus X (MVX), found in A. bisporus [5,6,7], Lentinula edodes spherical virus, found in Lentinula edodes [8], and Flammulina velutipes browning virus (FvBV), found in Flammulina filiformis [9, 10].
Viral metagenomics is a new virus detection method based on the rapid development of high-throughput sequencing technology. The genomes of fungal viruses are double-stranded RNA (dsRNA), positive-sense single-stranded RNA (+ssRNA), negative-sense single-stranded RNA (−ssRNA), or single-stranded DNA (ssDNA) [10,11,12,13,14,15]. The genomes of ssRNA and dsRNA viruses have been reported in edible mushrooms [16], but negative-stranded RNA viruses are rarely reported [17]. Mymonaviridae is a family of negative-strand mycoviruses belonging to the order Mononegavirales [18]. In general, viruses of the family Mymonaviridae have a linear single-segment RNA genome of about 10 kb, and the host range mainly includes fungi [18, 19]. The genome has no poly(A) tail structure at the 3' end, and the virions are filamentous enveloped particles with a diameter of 25–50 nm and a length of about 1,000 nm [18, 19].
Auricularia heimuer is widely cultivated in China and used as a dietary supplement due to its medicinal properties. However, the effect of viral infections on the quality and yield of this fungus has not yet been investigated. In this study, strains of A. heimuer collected from local factories were subjected to RNA sequencing and found to be infected with a virus, which was shown by phylogenetic analysis to be a new member of the family Mymonaviridae. This is the first negative-strand RNA virus and the third virus discovered in A. heimuer [20, 21].
A. heimuer strain CCMJ1222 was collected from the Engineering Research Center of Edible and Medicinal Fungi, Ministry of Education, Jilin Agricultural University. The strain was cultured on potato dextrose agar for seven days in the dark at 25℃. Fungal total RNA was extracted using an RNAiso Kit according to the manufacturer's instructions (Takara, Dalian, China). The total RNA samples were treated with DNase I (Takara, Dalian, China), and their integrity was evaluated by agarose gel electrophoresis and spectrophotometry using a NanoDrop 1000 instrument (Thermo Scientific, Wilmington, DE, USA). Library construction and deep sequencing were performed by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China).
After deep sequencing, adapter sequences and low-quality reads were removed from the raw reads of each library using Trimmomatic (version 0.36) [22]. The clean reads were mapped to the A. heimuer genome [23] using HISAT2 (version 2.1.0) [24]. The unmapped reads were assembled using Trinity (version 2.5.0) by de novo assembly [25]. The assembled contigs were compared to the NCBI nonredundant protein database using BLASTn and BLASTX (with significant e-value ≤ 1e−5) to identify virus-related contigs. The virus-related contigs that matched to the same reference sequence were merged into longer contigs using DNAMAN software (version 8.0; Lynnon Biosoft, Quebec, Canada).
Specific primers were designed based on the virus-related contigs and used for RT-PCR assays to identify viral sequences in the A. heimuer strains. First-strand cDNA was synthesized using a cDNA Synthesis SuperMix kit (TransGen, Beijing, China), using total RNA from A. heimuer obtained previously as a template. The 18S rRNA gene was used as an internal reference gene to analyze the synthesized cDNA [26]. Based on the verified sequences, we identified a novel virus, which we named “Auricularia heimuer negative-stranded RNA virus 1 DW” (AhNsRV1-DW). AhNsRV1-DW-specific primers were used to identify the target strain by PCR (Supplementary Table S1).
To verify the integrity of the virus genome, nested PCR was carried out with multiple sets of overlapping primers using the RACE method [26,27,28,29]. PCR products were separated by agarose gel electrophoresis, recovered using a Gel Extraction Kit (Axygen, NY, USA), and cloned into the vector pMD18-T (TaKaRa, Dalian, China). The primer pair M13-47/M13-48 (CGCCAGGGTTTTCCCAGTCACGAC/AGCGGATAACAATTTCACACAGGA) and specific primers were used for PCR detection of cloned sequences in bacteria cultured in liquid medium (Supplementary Table S1). The liquid culture medium containing the target bacteria was sent to Wuhan Quintarabio Biotechnology Co., Ltd. for sequencing. Each sample was sequenced three times to obtain high-quality sequences and avoid false-positive PCR results. Finally, the full-length cDNA sequence of the virus in A. heimuer strain CCMJ1222 was obtained through SnapGene splicing.
Based on the whole genome sequence, 3–4 specific primers were designed using the NCBI online software Primer Blast to verify the accuracy of the full-length sequence, and the RACE method was used to verify the terminal sequences. KOD high-fidelity enzyme was used for amplification. The PCR reaction was performed according to the instruction manual. A pTOPO-Blunt Kit (Aidlab, Beijing, China) was used for blunt-end cloning according to the kit instructions.
Conserved domains in AhNsRV1 were identified using the online software MOTIF (https://www.genome.jp/tools/motif/. The BLAST program at NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to search for homologous sequences. A phylogenetic tree was constructed by the maximum-likelihood method. An amino acid sequence alignment was performed using MAFFT and ClustalX software [30, 31], and the phylogenetic tree was visualized and edited using MEGA X.
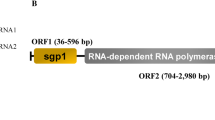
The genome of AhNsRV1 is 11,441 nt in length and has a GC content of 59.5%. The genome contains six ORFs with no overlaps (Fig. 1A). There is no cap structure at the 5′ end and no poly(A) structure at the 3′ end. The virus contains a 5′ UTR of 57 nt and a 3′ UTR of 296 nt. The 3'-UTR and 5′-UTR sequences had obvious complementary characteristics and were similar sequences (Fig. 1B). The genome structure of AhNsRV1 is similar to that reported for Lentinula edodes negative-stranded RNA virus 1 (LeNsRV1) [17] of the family Mymonaviridae (Fig. 1A). The spacer region has a series of conserved sequence elements rich in A/U (Fig. 1C). The largest ORF in AhNsRV1 is ORF VI. ORF VI is 5,908 nt long and encodes a putative L protein (RNA-dependent RNA polymerase [RdRP]) with 30.61% sequence identity to the RdRp of a virus from Golovinomyces cichoracearum. The L protein has 1,969 amino acid residues with a calculated molecular weight of 220.59 kDa and an isoelectric point of 6.64. The protein contains the conserved motif GxxTx (n) HR, which plays a key role in forming the mRNA cap structure [32, 33]. The other ORFs encode hypothetical proteins for which no homologs were found in the NCBI database. A BLAST search showed that the nucleotide sequence of the AhNsRV1 genome is 36.62% identical to that of Bondarzewia berkeleyi negative-strand RNA virus 1 and 34.94% identical to that of Armillaria mellea negative-strand RNA virus 1. The virus has been deposited in GenBank with the accession number MT259204.

Structure and characteristics of the AhNsRV1 genome. (A) Comparison of the size and structure of the AhNsRV1 and LeNsRV1 genomes. (B) The 3′ UTR and 5′ UTR reverse complementary sequences of AhNsRV1 are very similar to each other, and identical amino acids are highlighted in yellow. (C) Multiple alignment of the sequence of the intergenic regions. The sequence direction is 3′–5′
Twelve representative viruses belonging to four families of the order Mononegavirales were selected for multiple alignment of their RdRp sequences, which revealed the presence of eight conserved motifs (Supplementary Fig. S1). Phylogenetic analysis based on this alignment showed that AhNsRV1 and some members of the family Mymonaviridae clustered into a single branch of the phylogenetic tree (Fig. 2). In terms of genomic structure, AhNsRV1 is similar to Lentinus edodes negative-strand virus 1 (LeNsRV1) of the family Mymonaviridae, which infects the edible fungus Lentinus edodes. Therefore, AhNsRV1 is a new member of the family Mymonaviridae. Based on our analysis, we suggest the assignment of AhNsRV1 to the genus Auricularimonavirus in the family Mymonaviridae [18].

RAxML phylogenetic tree constructed based on amino acid sequences of the conserved domain of RdRp of AhNsRV1 and members of four families of the order Mononegavirales. The position of AhNsRV1 is shown in red
In this study, we isolated a novel mycovirus with a single-stranded negative-strand RNA genome from A. heimuer strain CCMJ1222. To the best of our knowledge, this is the first report of a negative-stranded RNA virus in A. heimuer. This study provides new information about negative-stranded RNA viruses in fungi.
References
Pearson MN, Beever RE, Boine B et al (2009) Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol Plant Pathol 10:115–128. https://doi.org/10.1111/j.1364-3703.2008.00503.x
Verma VC, Gange AC (2014) Entomopathogenic and nematophagous fungal endophytes. Adv Endophytic Res. https://doi.org/10.1007/978-81-322-1575-2_4
Ghabrial SA, Castón JR, Jiang D et al (2015) 50-plus years of fungal viruses. Virology 479–480:356–368. https://doi.org/10.1016/j.virol.2015.02.034
Sinden JW, Hauser E (1950) Report on two new mushroom diseases. Mushroom Sci 1(1):96–100
Tavantzis SM, Romaine CP, Smith SH (1980) Purification and partial characterization of a bacilliform virus from Agaricus bisporus: A single-stranded RNA mycovirus. Virology 105(1):94–102. https://doi.org/10.1016/0042-6822(80)90159-2
Grogan HM, Adie BAT, Gaze RH et al (2003) Double-stranded RNA elements associated with the MVX disease of Agaricus bisporus. Mycol Res 107(2):147–154. https://doi.org/10.1017/s0953756203007202
Elibuyuk IO, Bostan H (2010) Detection of a virus disease on white button mushroom (Agaricus bisporus) in Ankara, Turkey. Int J Agric Biol 12:597–600. https://doi.org/10.1016/j.compag.2010.03.005
Won HK, Park SJ, Kim DK et al (2013) Isolation and characterization of a mycovirus in Lentinula edodes. J Microbiol 51(1):118–122. https://doi.org/10.1007/s12275-013-2351-2
Wang M, Liu X, Dai Y et al (2018) Phylogeny and species delimitation of Flammulina: taxonomic status of winter mushroom in East Asia and a new European species identified using an integrated approach. Mycol Prog 17:1013–1030. https://doi.org/10.1007/s11557-018-1409-2
Magae Y, Sunagawa M (2010) Characterization of a mycovirus associated with the brown discoloration of edible mushroom, Flammulina velutipes. Virol J 7(1):342. https://doi.org/10.1186/1743-422X-7-342
Yu X, Li B, Fu Y et al (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci USA 107(18):8387–8392. https://doi.org/10.1073/pnas.0913535107
Yu HJ, Lim D, Lee HS (2003) Characterization of a novel single-stranded RNA mycovirus in Pleurotus ostreatus. Virology 314(1):9–15. https://doi.org/10.1016/s0042-6822(03)00382-9
Yu X, Li B, Fu Y et al (2013) Extracellular transmission of a DNA mycovirus and its use as a natural fungicide. Proc Natl Acad Sci USA 110(4):1452–1457. https://doi.org/10.1073/pnas.1213755110
Liu L, Xie J, Cheng J et al (2014) Fungal negative-stranded RNA virus that is related to bornaviruses and nyaviruses. Proc Natl Acad Sci USA 111(33):12205–12210. https://doi.org/10.1073/pnas.1401786111
Revill PA, Wright PJ (1997) RT-PCR detection of dsRNAs associated with La France disease of the cultivated mushroom Agaricus bisporus (Lange) Imbach. J Virol Methods 63(1–2):17–26. https://doi.org/10.1016/s0166-0934(96)02113-1
Chang Y, Chen J, Chang K et al (2019) Cloning and expression of the lectin gene from the mushroom Agrocybe aegerita and the activities of recombinant lectin in the resistance of shrimp white spot syndrome virus infection. Dev Comp Immunol 90(02):1–9. https://doi.org/10.1016/j.dci.2018.07.020
Lin YH, Fujita M, Chiba S et al (2019) Two novel fungal negative-strand RNA viruses related to mymonaviruses and phenuiviruses in the shiitake mushroom (Lentinula edodes). Virology 533:125–136. https://doi.org/10.1016/j.virol.2019.05.008
Kuhn JH, Adkins S, Agwanda BR et al (2021) 2021 Taxonomic update of phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch Virol 166(12):3513–3566. https://doi.org/10.1007/s00705-021-05143-6
Jiāng D, Ayllón MA, Marzano SL (2019) ICTV Report Consortium. ICTV virus taxonomy profile: Mymonaviridae. J Gen Virol 100:1343–1344. https://doi.org/10.1099/jgv.0.001301
Li X, Xie J, Hai D et al (2020) Molecular characteristics of a novel ssRNA virus isolated from Auricularia heimuer in China. Arch Virol 165(6):1495–1499. https://doi.org/10.1007/s00705-020-04615-5
Li X, Sui K, Xie J, Hai D et al (2020) Molecular characterization of a novel fusarivirus infecting the edible fungus Auricularia heimuer. Arch Virol 165(11):2689–2693. https://doi.org/10.1007/s00705-020-04781-6
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Yuan Y, Wu F, Si J et al (2019) Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 111:50–58. https://doi.org/10.1016/j.ygeno.2017.12.013
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. https://doi.org/10.1038/nmeth.3317
Haas BJ, Papanicolaou A, Yassour M et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. https://doi.org/10.1038/nprot.2013.084
Fan X, Zhou Y, Xiao Y et al (2014) Cloning, expression and phylogenetic analysis of a divergent laccase multigene family in Auricularia auricula-judae. Microbiol Res 169(5–6):453–462. https://doi.org/10.1016/j.micres.2013.08.004
Chiba S, Salaipeth L, Lin YH et al (2009) A novel bipartite double-stranded RNA mycovirus from the white root rot fungus Rosellinia necatrix: molecular and biological characterization, taxonomic considerations, and potential for biological control. J Virol 83(24):12801–12812. https://doi.org/10.1128/JVI.01830-09
Darissa O, Willingmann P, Adam G (2010) Optimized approaches for the sequence determination of double-stranded RNA templates. J Virol Methods 169(2):397–403. https://doi.org/10.1016/j.jviromet.2010.08.013
Marzano SL, Nelson BD, Ajayi-Oyetunde O et al (2016) Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J Virol 90:6846–6863. https://doi.org/10.1128/JVI.00357-16
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Kazutaka K, Standley DM (2016) A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 32:1933–1942. https://doi.org/10.1093/bioinformatics/btw108
Longdon B, Murray GG, Palmer WJ et al (2015) The evolution, diversity, and host associations of rhabdoviruses. Virus Evol 1(1):vev014. https://doi.org/10.1093/ve/vev014
Ogino T (2013) In vitro capping and transcription of rhabdoviruses. Methods 59(2):188–198. https://doi.org/10.1016/j.ymeth.2012.05.013
Acknowledgments
We gratefully acknowledge Qingcheng Liu and Guosheng Zhu from Guizhou Key Laboratory of Edible Fungi Breeding, Guizhou Academy of Agricultural Sciences, for providing us with their help. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (2021YFD1600401) and the Program of Introducing Talents of Discipline to Universities (No. D17014).
Author information
Authors and Affiliations
Contributions
Conceptualization: YL, CL, and YF. Data curation: XL and SL. Form analysis: XL and FLS. Funding acquisition: YL and CL. Methodology: XL. Software: XL. Supervision: YL, CL, and YF. Validation: XL, FLS, QL, and GZ. Writing–original draft: XL. Writing–review and editing: XL, FLS, CL, QL, XH, and GZ. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Massimo Turina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Liu, Q., Li, S. et al. A novel fungal negative-stranded RNA virus related to mymonaviruses in Auricularia heimuer. Arch Virol 167, 2223–2227 (2022). https://doi.org/10.1007/s00705-022-05540-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05540-5