
Abstract
The spotted lanternfly (Lycorma delicatula) is an invasive pest that causes serious economic losses in fruit and wood production. Here, we identified a novel iflavirus named "Lycorma delicatula iflavirus 1" (LDIV1), in a spotted lanternfly. The full genome sequence of LDIV1 is 10,222 nt in length and encodes a polyprotein containing a picornavirus capsid-protein-domain-like domain, a cricket paralysis virus capsid superfamily domain, an RNA helicase domain, a peptidase C3 superfamily domain, and an RNA-dependent RNA polymerase (RdRp) domain. LDIV1 replicates in the host insect and activates small interfering RNA (siRNA)-based host antiviral immunity. Phylogenetic analysis demonstrated that LDIV1 is most closely related to an unspecified member of the order Picornavirales, with 61.7% sequence identity in the RdRp region and 57.6% sequence identity in the coat protein region, and thus meets the demarcation criteria for new species in the genus Iflavirus. To the best of our knowledge, LDIV1 is the first virus discovered in L. delicatula.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The spotted lanternfly Lycorma delicatula (Hemiptera: Fulgoridae) is a sap-feeding insect native to China. It is highly polyphagous and feeds on more than 70 plant species, including apples and grapes [1]. In recent years, owing to the increasing frequency of international trade, the distribution range of L. delicatula has been expanding from China to Japan, South Korea, and the United States, causing serious damage to the global fruit and wood industry [2, 3]. The current management strategies for L. delicatula mainly rely on pesticide application, which is often harmful to its natural enemies and can result in pest resurgence. In 2019, a high-quality reference genome sequence of L. delicatula became available [4], which provided a valuable resource for investigating this invasive pest. However, as far as we know, there is little or no information about the viruses infecting L. delicatula.
Iflaviridae is a family of positive-stranded, non-enveloped RNA viruses with a small linear genome (approximately 9-11 kilobases in length) [5]. According to the latest ICTV Online Report (http://ictv.global/report/), the family Iflaviridae includes only 16 species in a single genus, Iflavirus. Genome architecture analysis has demonstrated that iflaviruses contain a single open reading frame (ORF), which is directly translated into a polyprotein, followed by posttranslational processing to generate the non-structural and structural proteins [5]. All viruses in the family Iflaviridae identified so far are restricted to infecting invertebrates, usually insects [6,7,8]. It has been reported that some iflaviruses are pathogenic for insects, causing premature mortality, behavioural changes, and developmental abnormalities in their hosts and thus can potentially be used as agents for the biological control of specific insects [9, 10].
The L. delicatula analyzed in this study was collected from a tree of heaven, Ailanthus altissima, in Xiangshan (29° N, 122° E), Zhejiang, China in 2020. Total RNA was extracted from the whole insect using a TRIzol Total RNA Isolation Kit (Takara, Dalian, China). The RNA sample was then sent to Novogene (Beijing, China) for transcriptomic sequencing as described previously [11]. Later, the raw output data were filtered, and the clean data were assembled using Trinity (version 2.8.5) [12] with default parameters. To confirm the insect species, the assembled contigs were compared with cytochrome oxidase subunit 1 (COI) from the Barcode of Life Data (BOLD) system [13], and the lanternfly was identified as L. delicatula, with a COI sequence identical to the reference sequence (accession number MN607209).
To identify the potential iflavirus, the assembled contigs were compared with a locally generated viral database containing sequences from all members of the genus Iflavirus. As a result, an ifla-like viral contig, representing an almost complete viral genome sequence, was detected. The viral contig was confirmed by reverse transcription PCR (RT-PCR), and the full genome sequence of the ifla-like virus was successfully obtained by rapid amplification of cDNA ends (RACE) using a SMARTer® RACE 5′/3′ Kit (Takara). The primers are listed in Supplementary Table S1. The new virus discovered in L. delicatula was named "Lycorma delicatula iflavirus 1" (LDIV1), and its complete genome sequence was submitted to the GenBank database (accession number OM728531).
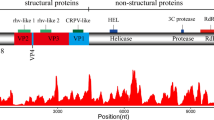
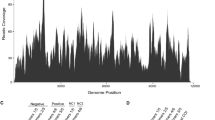
Excluding the polyA tail, the whole genome sequence of LDIV1 is 10,222 nt in length and contains a large ORF (nt 573 to 9992), a 5′-UTR (572 nt) and a 3′-UTR (230 nt). The large ORF encodes a 3,140-aa polyprotein. Analysis using InterPro Scan revealed typical domains of iflaviruses [14], including a picornavirus capsid-protein-domain-like (rhv-like) domain (accession no. IPR033703, E-value: 1.1 × 10-24), a cricket paralysis virus (CRPV) capsid superfamily domain (IPR014872, 1.5 × 10-4), an RNA helicase (Hel) domain (IPR000605, 4.6 × 10-20), a peptidase C3 superfamily (peptidase) domain (IPR009003, 2.3 ×10-9) and an RNA-dependent RNA polymerase (RdRp) domain (IPR043502, 1.5 × 10-95) (Fig. 1A). Moreover, the abundance and coverage of LDIV1 were assessed by rearranging RNA-seq reads into a reconstructed complete genome sequence of LDIV1 using Bowtie2 with zero mismatches [15]. A total of 4,768 reads were perfectly mapped to LDIV1 genome sequences, accounting for 0.02% of the total RNA-seq reads (Fig. 1B). Currently, one RNA-seq dataset of L. delicatula (accession no. SRR5134712) is available in the NCBI SRA repository. To investigate the prevalence of LDIV1, RNA-seq reads of SRR5134712 were assembled into an LDIV1 genome sequence. As shown in Supplementary Fig. S1, only 84 reads (<0.0001% of the total RNA-seq reads) mapped to the LDIV1 genome. These results indicated that LDIV1 was prevalent in L. delicatula, but the viral abundance differed among individuals.

(A) Schematic diagram of the LDIV1 genome structure. (B) Transcriptome raw read coverage. rhv-like, picornavirus capsid-protein-domain-like; CRPV, cricket paralysis virus capsid superfamily domain; Hel, RNA helicase domain; peptidase, peptidase C3 superfamily domain; RdRp, RNA-dependent RNA polymerase domain. (C) Maximum-likelihood phylogenetic tree based on the RNA-dependent RNA polymerase domain, constructed using 1000 bootstrap replicates. The scale bar represents percent divergence. Drosophila immigrans Nora virus and Spodoptera exigua virus were used as an outgroup.
The LDIV1 polyprotein showed the highest amino acid sequence identity to an unspecified member of the order Picornavirales (MT138389). The amino acid sequence identity value in the CP is one of the criteria used to define new iflaviruses. The percentage identity values of 57.6% in the CP and 61.7% in the RdRp when compared with its closest relative indicate that this virus meets the demarcation criteria for a new species in the genus Iflavirus, with the sequence identity in the CP being less than 90% at the amino acid level [16]. It is worth mentioning that, although the Picornavirales member to which LDIV1 was found to be related was detected in the anus of a bird by metagenomic analysis, BLAST results showed that its close relatives are insect viruses. It is therefore likely that it was an insect virus that was part of the diet of the bird. To determine the taxonomic status of LDIV1, the RdRp amino acid sequence was aligned to those of the previously reported iflaviruses, using MAFFT (version 7.450) [17]. The substitution model was evaluated using ModelTest-NG, and a maximum-likelihood phylogenetic tree was constructed in RAxML-NG (version 0.9.0), with 1000 bootstrap replicates [18]. Phylogenetic analysis showed LDIV1 to be most closely related to "Picornavirales sp." and Nilaparvata lugens honeydew virus 3 (Fig. 1C).
To obtain information about small interfering RNA (siRNA)-based host antiviral immunity, a small RNA (sRNA) library of L. delicatula was also sequenced, and virus-derived siRNA (vsiRNAs) were comprehensively characterized as described previously [19]. The sRNA library was prepared using a Illumina TruSeq Small RNA Sample Preparation Kit (Illumina, USA) and sequenced on an Illumina HiSeq 2500 platform. The sRNA reads were processed by removing adapters, low-quality sequences, and unwanted sequences using Trimmomatic (Version 3.90) [20], and sRNAs with a length of 18-30 nt were extracted. A total of 28,617 sRNA reads (8957 unique) were successfully mapped to the assembled genome sequence of LDIV1, accounting for 0.16% (0.50% unique) of the whole sRNA library (Supplementary Table S2). siRNA-based RNA silencing is usually associated with the accumulation of vsiRNAs in a sequence-specific manner. For example, a 21-nt vsiRNA length peak was observed in Diaphorina citri and Scathophaga furcate [19, 21], while a 22-nt vsiRNA peak were observed in Bemisia tabaci and Astegopteryx formosana [22, 23]. In this study, the majority of LDIV1-derived vsiRNAs were 22-23 nt in length (Fig. 2A) and were almost equally derived from the sense and antisense strands of the virus genome (Fig. 2B). The presence of these LDIV1-derived vsiRNAs strongly suggested that the RNAi antiviral pathway of L. delicatula was activated in response to LDIV1 infection.

Analysis of Lycorma delicatula iflavirus 1 (LDIV1)-derived small interfering RNAs (siRNA). (A) The size distribution of LDIV1-derived siRNAs with a length of 18-30 nt. (B) Distribution of LDIV1-derived siRNA on the viral genome. Black represents sRNAs derived from the antisense strand of the genome (minus), and red represents sRNAs derived from the sense strand of the genome (plus).
In conclusion, we have determined the full genome sequence of LDIV1 from an invasive pest, L. delicatula. LDIV1 is a member of the genus Iflavirus that is evolutionarily related to a previously reported member of the order Picornavirales. LDIV1 replicates in host insects and activates the RNAi antiviral pathway. In the future, it will be interesting to determine the influence of LDIV1 on its host and investigate the potential value of this virus for biological control of agricultural pests.
Data availability
Raw reads generated by transcriptomic and sRNA sequencing have been deposited in the Genome Warehouse of the National Genomics Data Center (https://bigd.big.ac.cn/gwh) under accession number PRJCA009021.
References
Avanesyan A, Lamp WO (2020) Use of molecular gut content analysis to decipher the range of food plants of the invasive spotted lanternfly, Lycorma delicatula. Insects 11(4):215
Dara SK, Barringer L, Arthurs SP (2015) Lycorma delicatula (Hemiptera: Fulgoridae): a new invasive pest in the United States. J Integrated Pest Manag 6(1):20
Han JM, Kim H, Lim EJ, Lee S, Kwon YJ, Cho S (2008) Lycorma delicatula (Hemiptera: Auchenorrhyncha: Fulgoridae: Aphaeninae) finally, but suddenly arrived in Korea. Entomol Res 38(4):281–286
Kingan SB, Urban J, Lambert CC, Baybayan P, Childers AK, Coates B, Scheffler B, Hackett K, Korlach J, Geib SM (2019) A high-quality genome assembly from a single, field-collected spotted lanternfly (Lycorma delicatula) using the PacBio Sequel II system. GigaScience 8(10):122
van Oers MM (2010) Genomics and biology of Iflaviruses. Insect Vrol 231–250
Wu N, Zhang P, Liu W, Cao M, Wang X (2018) Sequence analysis and genomic organization of a new insect iflavirus, Sogatella furcifera honeydew virus 1. Adv Virol 163(7):2001–2003
Wang H, Liu Y, Liu W, Cao M, Wang X (2019) Full genome sequence of a novel iflavirus from the leafhopper Psammotettix alienus. Adv Virol 164(1):309–311
Ye ZX, Li YH, Chen JP, Zhang CX, Li JM (2021) Complete genome analysis of a novel iflavirus from a leaf beetle, Aulacophora lewisii. Arch Virol 166(1):309–312
Ryabov EV, Wood GR, Fannon JM, Moore JD, Bull JC, Chandler D, Mead A, Burroughs N, Evans DJ (2014) A virulent strain of deformed wing virus (DWV) of honeybees (Apis mellifera) prevails after Varroa destructor-mediated, or in vitro, transmission. PLoS Pathog 10(6):e1004230
Bailey L, Gibbs A, Woods R (1964) Sacbrood virus of the larval honey bee (Apis mellifera Linnaeus). Virology 23(3):425–429
Huang HJ, Ye ZX, Lu G, Zhang CX, Chen JP, Li JM (2021) Identification of salivary proteins in the whitefly Bemisia tabaci by transcriptomic and LC-MS/MS analyses. Insect Sci 28(5):1369–1381
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Sujeevan R, Hebert P, Diego F (2013) A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS ONE 8(7):e66213
Blum M, Chang H-Y, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA, Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I, Marchler-Bauer A, Mi H, Natale DA, Necci M, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A, Finn RD (2020) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49(D1):D344–D354
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359
Valles SM, Chen Y, Firth AE, Guerin DMA, Hashimoto Y, Herrero S, de Miranda JR, Ryabov E, Ictv Report C (2017) ICTV virus taxonomy profile: Iflaviridae. J Gen Virol 98(4):527–528
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30(4):772–780
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35(21):4453–4455
Lu G, Ye ZX, He YJ, Zhang Y, Wang X, Huang HJ, Zhuo JC, Sun ZT, Yan F, Chen JP, Zhang CX, Li JM (2020) Discovery of two novel negeviruses in a dungfly collected from the Arctic. Viruses 12(7):692
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Nigg JC, Kuo YW, Falka BW (2020) Endogenous viral element-derived piwi-interacting RNAs (piRNAs) are not required for production of ping-pong-dependent piRNAs from Diaphorina citri densovirus. MBio 11(5):e02209-e2220
Zhang XD, Lu G, Ye ZX, Chen JP, Zhang CX, Li JM (2022) Complete genome analysis of a nege-like virus in aphids (Astegopteryx formosana). Adv Virol 167(1):267–270
Huang HJ, Ye ZX, Wang X, Yan XT, Zhang Y, He YJ, Qi YH, Zhang XD, Zhuo JC, Lu G, Lu JB, Mao QZ, Sun ZT, Yan F, Chen JP, Zhang CX, Li JM (2021) Diversity and infectivity of the RNA virome among different cryptic species of an agriculturally important insect vector: whitefly Bemisia tabaci. npj Biofilms Microbiomes 7(1):43
Funding
This research was supported by the National Natural Science Foundation of China (U20A2036) and the Ningbo Science and Technology Innovation 2025 Major Project (2019B10004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Simona Abba' .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
705_2022_5503_MOESM1_ESM.tif
Assembly of RNA-seq reads to obtain the Lycorma delicatula iflavirus 1 (LDIV1) genome sequence. The RNA-seq dataset of L. delicatula (accession number: SRR5134712) was retrieved from NCBI SRA repository and mapped to the LDIV1 genome. (TIF 3480 KB)
Rights and permissions
About this article

Cite this article
Wang, YZ., Ye, ZX., Chen, J. et al. Complete genome analysis of a novel iflavirus from the spotted lanternfly Lycorma delicatula. Arch Virol 167, 2079–2083 (2022). https://doi.org/10.1007/s00705-022-05503-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05503-w