
Abstract
A novel enamovirus was identified in common bean plants with disease symptoms. Its genome of 5,781 nucleotides (nt) contains five open reading frames. This virus and other members of the genus Enamovirus share 50.4-68.4% nucleotide sequence identity in the complete genome and 19.9-51.9% amino acid sequence identity in the P0 protein, 24.9-52.5% in P1, 33.4-62.9% in P1-P2, 30.6-81.1% in P3, and 32.3-74.2% in P3-P5. Phylogenetic analysis showed that the virus is most closely related to alfalfa enamovirus 1 and pea enation mosaic virus 1 in the genus Enamovirus of the family Solemoviridae. These results suggest that this virus, tentatively named “bean enamovirus 1”, should be classified as a member of a new species in the genus Enamovirus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Common bean (Phaseolus vulgaris L.), also called French bean, is a herbaceous annual crop belonging to the family Fabaceae. It is native to Mesoamerica and was introduced into China in the 15th century [1]. Young pods of common bean are used as vegetables, and the dried seeds are one of the major staple foods in countries of Africa, Latin America, the Caribbean, and Asia. According to statistics from the Food and Agriculture Organization of the United Nations (FAO) in 2018, China is the fifth-largest producer of common bean, with an annual planting area of about 100 million hm2 and an output of nearly 1.3 million tons. However, common bean production is vulnerable to many diseases, including those caused by viruses. At least 43 viruses have been reported to infect common bean and cause a decline in production [2,3,4].
The genus Enamovirus was originally part of the family Luteoviridae, but recently, it was moved to the family Solemoviridae, which also includes the genera Polemovirus, Polerovirus, and Sobemovirus [5]. Enamoviruses have a single-stranded RNA genome of 5.7-6.0 kb in length that contains five open reading frames (ORF). They have a narrow host range and are transmitted by aphids. In this study, a novel enamovirus was identified in common bean and molecularly characterized. A survey of multiple crops showed that the virus infects several other legume crops.
In September 2020, common bean samples with symptoms of mild mottle were collected from fields in Kunming, Yunnan, China (Fig. 1A). To identify potential pathogens, five symptomatic samples were pooled for preparation of a total RNA sample that was subjected to high-throughput sequencing (HTS). The rRNA-depleted sample was used for construction of an RNA-seq library for sequencing on an Illumina HiSeq X Ten platform (Origingene Bio-pharm Technology, Shanghai, China). The RNA-seq data were analyzed using CLC Genomic Workbench 20.0 (QIAGEN). A total of 48,792,184 paired-end reads were obtained, and de novo assembly of the reads generated 138,508 contigs with more than 200 nucleotides (nt). A BLASTx search of the GenBank database revealed that about 0.1% of the total reads corresponded to bean yellow mosaic virus, tomato spotted wilt virus, or alfalfa mosaic virus, but there was an additional large contig of 5,374 nt with average coverage of 243.63 and the highest nt sequence identity of 72.3% to alfalfa enamovirus 1 (AEV-1, GenBank accession no. KU297983).

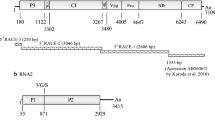
Symptoms on common bean (A), alfalfa (B), and vetch (C) infected with bean enamovirus 1 (BEnV-1) and a schematic representation of the genomic organization of BEnV-1 (D). The open reading frames (ORFs) are depicted by rectangles with different colors. The start and stop points of each ORF are labeled. Translation products are depicted as blue rectangles.
To determine the complete genome sequence of the novel enamovirus by HTS, six pairs of overlapping primers were designed based on the large contig covering most of the virus genome (Supplementary Table S1). Total RNA was extracted from individual plant samples using TRIpure Reagent (Bioteke Corporation, Beijing, China) and tested by RT-PCR for the presence of the new virus. An extract from an infected plant was used for amplification of fragments of the viral genome by RT-PCR. The PCR products were cloned into the vector pMD19-T (TaKaRa Biotechnology Co., Ltd, Dalian, China), and at least three clones were sequenced for each amplicon (BGI, Guangzhou, China). The 5’- and 3’-terminal sequences were amplified using a SMARTer®RACE 5’/3’ Kit as described by the manufacturer (TaKaRa Biotechnology, Dalian, China). The RT-PCR amplification, cloning, and sequence analysis were conducted as described by Lan et al. [6].
The complete genome of the new enamovirus consists of 5,781 nt (GenBank accession no. MZ361924) and contains five open reading frames (ORF). Its genomic organization is typical of members of the genus Enamovirus (Fig. 1C). The 5’ and 3’ untranslated regions (UTR) are 174 nt and 233 nt long, respectively, and the intergenic region (IR) between ORFs 2 and 3 is 188 nt in length. ORF0, starting at nt 175 and ending at nt 1,086, encodes a putative P0 protein of 303 amino acid (aa) residues that contains the sequence 213LPxxL217, the putative F-box-like motif, which is conserved among enamoviruses [7]. ORF1 is predicted to be expressed by a ribosomal leaky scanning mechanism, encoding a putative P1 protein of 766 aa residues. The conserved sequence 390H(x29)D(x67)GxSG591 of the S39 serine protease is present in this region [8]. ORF2 is predicted to be translated by a -1 ribosomal frameshift at a putative slippery heptamer, 2039GGGAAAT2045, within ORF1 to produce a fusion protein of P1-P2 with a length of 1188 aa residues. P1-P2 might be involved in replication of enamoviruses, as it contains the highly conserved motif 1,085GDD1,087 in the viral RNA-dependent RNA polymerase (RdRp) [8]. After a short IR, ORF3 starts at nt 4,022 and ends at nt 4,594, encoding a coat protein (CP) of 190 aa residues. ORF5 is predicted to be expressed by a putative in-frame readthrough from ORF3, producing a fusion protein of 508 aa residues (P3-P5). P3-P5 is thought to be an aphid-transmission subunit in members of the genus Enamovirus [9].
Pairwise comparisons of the genome sequences and the predicted aa sequences of the individual proteins of this virus and other members of the genus were conducted, and the results show that this new virus shares 50.4-68.4% nt sequence identity with enamoviruses at the whole-genome level and 19.9-51.9% amino acid sequence identity in P0, 24.9-52.5% in P1, 33.4-62.9% in P1-P2, 30.6-81.1% in P3, and 32.3-74.2% in P3-P5 (Supplementary Table S2). These values are below the species demarcation threshold of 10% aa sequence identity in any gene product for the genus Enamovirus [10]. It is worth noting that the CP is the most conserved protein, whereas ORF0 is the most divergent among the enamoviruses. A maximum-likelihood tree based on deduced P1-P2 fusion protein sequences of this virus and members of the genera Enamovirus, Polemovirus, Sobemovirus, and Polerovirus of the family Solemoviridae places the new virus together with AEV-1 [11] and pea enation mosaic virus 1 (PEMV-1) [12] in a legume clade (Fig. 2). The results of these analyses suggest that the new virus, tentatively named "bean enamovirus 1" (BEnV-1), should be considered a new member of the genus Enamovirus.

Maximum-likelihood tree based on the deduced fusion protein sequences of P1-P2 of BEnV-1 and members of the genera Enamovirus, Sobemovirus, Polemovirus, and Polerovirus. Bootstrap analysis was applied using 1000 bootstrap replicates. A solid triangle indicates the BEnV-1 characterized in this study.
To investigate the distribution and potential natural host species of BEnV-1 in the field, a total of 378 leaf samples were collected from different crops and weeds with or without symptoms in Yunnan. RT-PCR was performed using the specific primers BEnVdF and BEnVdR (Supplementary Table S1), and five out of 25 vetch plants (21.7%), three out of 10 alfalfa plants (30.0%), and five out of 59 common bean plants (8.5%) were found to be infected with BEnV-1. The main symptom associated with BEnV-1 in the three infected legume plants was mild mottle (Fig. 1A-C). The virus was not detected in cowpea, soybean, pea, faba bean, pepper, tomato, potato, cucurbit, cucumber, and passion fruit. Common bean and alfalfa are important food and forage crops throughout the world. The effects of BEnV-1 on its hosts still needs to be assessed.
References
Zhang J (2014) Study on the development and transmission history of Kidney bean. Agric Archaeol 4:218–229
Mwaipopo B, Nchimbi-Msolla S, Njau P, Tairo F, William M, Binagwa P, Kweka E, Kilango M, Mbanzibwa D (2017) Viruses infecting common bean (Phaseolus vulgaris L.) in Tanzania: a review on molecular characterization, detection and disease management options. Afr J Agric Res 12(8):1486–1500
Kamaal N, Akram M, Agnihotri AK (2015) Molecular evidence for the association of tomato leaf curl Gujarat virus with a leaf curl disease of Phaseolus vulgaris L. J Phytopathol 163(1):58–62
Liu Y, Li F, Li YY, Zhang SB, Gao XW, Xie Y, Yan F, Zhang AS, Dai LY, Cheng ZB, Ding M, Niu YB, Wang SJ, Che HY, Jiang T, Shi XB, He ZF, Wu YF, Zhang DY, Qing L, Yan WR, Yang XH, Tang YF, Zheng HY, Tang QJ, Zhang SB, Zhang DF, Cai L, Tao XR (2019) Identification, distribution and occurrence of viruses in the main vegetables of China. Scientia Agricultura Sinica 52(2):239–261
Sõmera M, Fargette D, Hébrard E, Sarmiento C (2021) Solemoviridae, ICTV https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/solemoviridae
Lan PX, He P, Zhang YK, Zhang S, Zhang ZB, Chen XJ, Tan ST, Luo HM, Cao MJ, Li F (2019) Molecular characterization of a novel potyvirus infecting noni. Arch Virol 164(12):3099–3102
Fusaro AF, Correa RL, Nakasugi K, Jackson C, Kawchuk L, Vaslin MFS, Waterhouse PM (2012) The Enamovirus P0 protein is a silencing suppressor which inhibits local and systemic RNA silencing through AGO1 degradation. Virol 426:178–187
Silva JME, Rwahnih MA, Blawid R, Nagata T, Fajardo TVM (2017) Discovery and molecular characterization of a novel enamovirus, grapevine enamovirus-1. Virus Genes 53(4):667–671
Demler SA, de Zoeten GA (1991) The nucleotide sequence and luteovirus-like nature of RNA 1 of an aphid non-transmissible strain of pea enation mosaic virus. J Gen Virol 72:1819–1834
Debat HJ, Bejerman N (2019) Novel bird’s-foot trefoil RNA viruses provide insights into a clade of legume-associated enamoviruses and rhabdoviruses. Arch Virol 164(5):1419–1426
Bejerman N, Giolitti F, Trucco V, Breuil S, Dietzgen RG, Lenardon S (2016) Complete genome sequence of a new enamovirus from Argentina infecting alfalfa plants showing dwarfism symptoms. Arch Virol 161(7):2029–2032
Palanga E, Martin DP, Galzi S, Zabre J, Bouda Z, Neya JB, Sawadogo M, Traore O, Peterschmitt M, Roumagnac P, Filloux D (2017) Complete genome sequences of cowpea polerovirus 1 and cowpea polerovirus 2 infecting cowpea plants in Burkina Faso. Arch Virol 162(7):1–4
Funding
This study was funded by the Yunnan Academician Expert Workstation (202005AF150040) and Program for Innovative Research Team (in Science and Technology) in University of Yunnan Province (Yunjiaoke 2014-22).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
As the corresponding authors, Ruhui Li and Fan Li declare no conflict of interest involving the authors of this paper.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Ralf Georg Dietzgen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lu, Rb., Lan, Px., Kang, Rj. et al. Genomic characterization of a new enamovirus infecting common bean. Arch Virol 167, 999–1002 (2022). https://doi.org/10.1007/s00705-022-05387-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05387-w