
Abstract
In the current pandemic of coronavirus disease 2019 (COVID-19), antiviral drugs are at the center of attention because of their critical role against severe acute respiratory disease syndrome coronavirus 2 (SARS-CoV-2). In addition to designing new antivirals against SARS-COV-2, a drug repurposing strategy is a practical approach for treating COVID-19. A brief insight about antivirals would help clinicians to choose the best medication for the treatment of COVID-19. In this review, we discuss both novel and repurposed investigational antivirals, focusing on in vitro, in vivo, and clinical trial studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by the novel severe acute respiratory disease syndrome coronavirus 2 (SARS-CoV-2) has become a health crisis with a high rate of morbidity and mortality. Despite the fact that numerous studies have been performed amid the pandemic, management of the disease has remained a major challenge. In addition to drug development projects, drug repurposing is another effective strategy to identify new clinical applications for existing drugs [97].
Remdesivir, the first Food and Drug Administration (FDA)-approved antiviral for treating COVID-19 patients, was initially developed against Ebola virus disease in 2016. Despite the early promising data from remdesivir trials, the World Health Organization (WHO) did not judge remdesivir to have a beneficial effect on survival rate [13, 34]. Therefore, there is an urgent need to develop additional antiviral drugs against COVID-19.
In this review, we discuss repurposed and investigational antivirals, focusing on in vitro, in vivo, and clinical trial studies (Tables 1, 2). To the best of our knowledge, this is the first review that categorizes these antivirals based on their mechanisms of action (Fig. 1).

Investigational antivirals and their mechanism of action during the viral life cycle
Virology
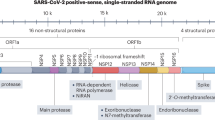
SARS-CoV-2 is an enveloped, positive-sense, single-stranded ribonucleic acid (RNA) virus belonging to the family Coronaviridae, subfamily Orthocoronavirinae, and genus Betacoronavirus [126]. Open reading frames in the RNA genome of SARS-CoV-2 encode viral structural and non-structural proteins [126].
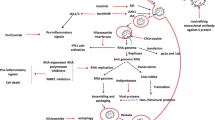
The process of viral infection is initiated by the binding of the viral surface spike (S) protein to two entry factors on the host cell: angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2) [99]. The viral S protein interacts separately with ACE2 and TMPRSS2 [99]. The expression levels of the viral S protein, ACE2, and TMPRSS2 affect the severity of disease in COVID-19 patients [99]. Moreover, the endosomal/lysosomal cysteine proteases cathepsin B and L (CTSB and CTSL) and furin protease are also involved in the viral entry [104]. After viral uncoating, viral replication, transcription, and translation occur to produce viral proteins (structural and non-structural). Finally, the assembled virus particles are released from the infected host cell via exocytosis, attacking the other cells and activating the innate immunity of the host [99].
The replication of SARS-CoV-2 mostly depends on the non-structural viral proteins, including the main protease (Mpro), the RNA-dependent RNA polymerase (RdRp), and the papain-like protease [8, 99]. Importantly, RdRp is a key target for newly discovered agents against SARS-CoV-2, since it is responsible for viral replication.
SARS-CoV-2 infection promotes the production of pro-inflammatory cytokines and inhibits the production of interferons (IFNs) [89]. Overproduction of pro-inflammatory cytokines and chemokines such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) leads to a cytokine storm, tissue injury, multiple organ failure, disseminated intravascular coagulation (DIC), and acute respiratory distress syndrome (ARDS) [89].
A brief insight into the SARS-CoV-2 cell cycle and pathogenesis would be helpful for drug discovery, as well as for a better understanding of COVID-19 complications [104]. Each step of virus trafficking and the immunological responses of the host could be considered a target for antiviral agents [60].
Investigational antivirals that block viral entry
Nitric oxide (NO)
The SARS-CoV-2-related hypercoagulable state and vascular complications play a leading role in the pathogenesis of ARDS [4]. Nitric oxide (NO) is a small multifunctional signaling molecule that improves vascular function [92]. NO is produced by the enzymatic conversion of L-arginine to L-citrulline. It is catalyzed by inducible NO synthase (iNOS) and requires O2, nicotinamide adenine dinucleotide phosphate (NADPH), calmodulin, and tetrahydrobiopterin as cofactors [90]. Endothelium-derived NO plays an essential role in vascular muscle relaxation, which results in vasodilation, reduced blood pressure, and increased local blood flow [52]. Moreover, NO prevents platelet aggregation, low-density lipoprotein (LDL) and cholesterol oxidation, and pro-inflammatory gene expression [81].
NO is a selective pulmonary vasodilator that has been approved by the FDA for hypoxic newborns with persistent pulmonary hypertension (at 10–80 ppm) [113]. Off-label uses of NO include treatment of pulmonary hypertension and ARDS [113]. In addition, NO is effective against several bacteria, protozoa, and viruses [37]. Laboratory data have indicated that NO has antiviral activity due to its ability to induce IFN-gamma expression in macrophages [90]. NO also regulates lymphocyte and monocyte function, providing signal transduction between innate and acquired immunity [81].
Akerstrom et al. reported that NO inhibits replication of SARS-CoV by two distinct mechanisms: reducing viral RNA replication and preventing S protein palmitoylation, thereby inhibiting binding of the viral S protein to the host ACE-2 receptor [2].
It is believed that NO may inhibit viral replication in the early stages, prevent viral spread, and aid in viral clearance and host recovery [90]. An in vitro study of SARS-CoV-infected Vero E6 cell lines indicated that treatment with NO donors increased the survival rate of the cells [64]. It has also been shown that NO inhalation improves oxygenation in patients with ARDS; however, it does not have a significant effect on the mortality rate [44]. Likewise, inhalation of NO at the dose of 20 ppm for 15–30 min significantly improves arterial oxygenation in COVID-19-induced moderate-to-severe ARDS [64, 74]. It also results in selective dilation of pulmonary vessels, improved arterial oxygenation, and reduced pulmonary hypertension. Ryan et al. have recommended the use of NO for treatment of pulmonary arterial hypertension in hospitalized patients with COVID-19 [37, 125].
A pilot study was carried out to determine the effectiveness of inhaled NO in addition to antiviral therapy in the treatment of patients with SARS-CoV. Of the 14 hospitalized intensive care unit (ICU) patients, six were assigned to the inhaled NO group and eight were assigned to the placebo group (age range: 19–63). There was no significant difference in the duration and severity of disease between the two groups. All of the patients received oxygen therapy in addition to antiviral therapy, which included ribavirin (0.5–1 mg/day) and methylprednisolone (40–160 mg/day). Patients in the treatment group received inhaled NO for at least 3 days with a dose of 30, 20, and 10 ppm on the first, second, and third day, respectively. After the fourth day, the dose of the inhaled NO was decreased gradually to 0 ppm. Data analysis demonstrated that low-dose inhaled NO decreased the mean oxygen delivery in patients from 6 to 2 L/min (p-value <0.05) and increased the mean oxygen saturation (SpO2) from 93% to 99% (p-value <0.05) in the treatment group; leading to a reduction in demand for ventilatory support in affected patients [23]. This study highlighted the vasodilatory effect of NO in addition to its effectiveness against SARS-CoV, since NO improved arterial oxygenation. The promising results with inhaled NO in patients infected by SARS-CoV suggest that it could also be effective in patients infected with SARS-CoV-2.
Akaberi et al. evaluated the in vitro antiviral effect of the NO donor S-nitroso-N-acetylpenicillamine on SARS-CoV-2 replication in Vero E6 cells. The results showed a dose-dependent reduction of viral replication due to inhibition of the viral cysteine protease [1]. Moreover, an in vitro study on human HCT-8 cells revealed a potential effect of high-dose inhaled NO (150–250 ppm, 30–40 min cycles, 2–5 cycles a day) against human coronavirus OC43 given either pre- or post-exposure [45]. In that study, infectivity of the virus decreased by 50% and 100% after 4 and 8 h of treatment, respectively [45].
A retrospective study was conducted to evaluate the efficacy of high-dose inhaled NO as rescue therapy in COVID-19 patients with respiratory distress. Five patients with severe COVID-19 were enrolled in the study, which was defined by an increased risk of acute hypoxemic respiratory failure with worsening symptoms despite oxygen therapy. All of the enrolled patients had comorbidities. Patients received NO at a concentration of 160 ppm for 30 min twice daily via a face mask. This study showed that NO therapy as adjuvant rescue therapy reduced hypoxemia and respiratory effort in severe COVID-19 patients. The median methemoglobin of patients at baseline and after 30 min of NO therapy was 0.3% (0.1–0.7%) and 2% (1.7–2.3%), respectively. Mean arterial pressure (MAP) and heart rate (HR) were not affected by the treatment. Moreover, the ratio of SpO2/FiO2 (oxygenation) and respiratory rate (RR) were stable [113]. This study revealed that NO therapy is a promising option for COVID-19 patients with respiratory failure. However, this study had some limitations, such as a small sample size and lack of a control group.
A monocentric pilot study was conducted by Bogate et al. to evaluate the effects of NO rescue therapy and almitrine in patients with COVID-19-related severe ARDS. Patients with reverse transcription polymerase chain reaction (RT-PCR)-confirmed COVID-19 with persistent severe hypoxemia, defined by PaO2/FiO2 < 150 mmHg, were enrolled in the study. The study exclusion criteria were age less than 18 years, acute cor pulmonale, pulmonary embolism, hyperlactatemia, hepatic insufficiency, and having receiving extracorporeal membrane oxygenation (ECMO) support. Ten patients with a median age of 60 years (52–72) were entered the study. All patients received inhaled NO (10 ppm) alone or NO plus almitrine (10 µg/kg/min) as a respiratory stimulant for 30 min. The primary endpoint of the study was variation in oxygenation, defined by an increase in the PaO2/FiO2 ratio of at least 20% or 20 mmHg from the baseline. The results showed an improvement in arterial oxygenation in 80% of the patients. The baseline median PaO2/FiO2 ratio when the patients were in a supine position was 102 (89–134), which was elevated to 124 (108–146) (p-value = 0.13) after treatment with NO and 180 (132–206) (p-value <0.01) after treatment with NO plus almitrine. These results show the beneficial effects of NO and almitrine combination therapy in the treatment of severe COVID-19. No correlation was observed between the success of NO plus almitrine combination therapy and prone position in severe COVID-19 patients [9].
At the time of writing of this manuscript, 15 ongoing clinical trials were being carried out to evaluate the efficacy of NO in COVID-19 patients. The studied dosage forms of NO are inhalation, lozenges, and solution. The trials target patients with mild-to-severe COVID-19 who need oxygen therapy or have pneumonia or bacterial infection due to COVID-19.
Preventive therapy with NO is also under investigation in ongoing clinical trials investigating the effects of NO on viral load, disease progression, mortality, respiratory failure, methemoglobinemia, and arterial oxygenation. Safety and adverse events of NO therapy are also being evaluated in these trials, with sample sizes ranging from 20 to 600.
Generally, inhaled NO is safe; however, some adverse events such as hemodynamic deterioration, methemoglobinemia, and rebound pulmonary hypertension have been reported, but these are reversible due to the short half-life of NO. Administration of NO may have some limitations. It needs active participation of patients during inhalation and needs to be administered appropriately to provide optimal efficacy [113].
NO seems to have the desired efficacy in patients with COVID-19. Given the safety and efficacy of NO in patients with COVID-19 and its availability and low cost, it is predicted to play an important role in managing COVID-19. The FDA has granted emergency use authorization (EUA) for NO therapy in COVID-19 patients.
Arbidol hydrochloride (umifenovir)
Arbidol, also known as umifenovir, is an indol-derived, broad-spectrum oral antiviral that is effective against both enveloped and non-enveloped viruses, including Ebola virus, Zika virus, and poliovirus [87]. Arbidol was first licensed in China and Russia to treat and prevent influenza A, influenza B, and other respiratory viral infections [109, 121]. It can also be used in the treatment of hepatitis C virus (HCV) [15, 87]. During the influenza pandemic, the prophylactic dose of arbidol in health workers was 200 mg daily for 10–18 days [42]. The recommended dose of arbidol for the treatment of influenza in Chinese guidelines is 200 mg, three times a day. The suggested mechanism of arbidol is blocking contact, adhesion, and fusion of the virus to the host cells and interfering with intracellular vesicle trafficking [127]. Moreover, arbidol inhibits viral RNA replication and induces an IFN/phagocyte-mediated immune response [55]. Arbidol has anti-inflammatory properties through increasing the IL-10 level and decreasing the levels of IL-1β, IL-6, IL-12, and TNF-alpha [71]. Wang et al. evaluated the efficacy of six anti-influenza agents against SARS-CoV-2 in Vero E6 cells. The results demonstrated that arbidol efficiently inhibits SARS-CoV-2 infection with a selectivity index of 7.73 [109].
An open-label randomized clinical trial (RCT) was carried out in Iran to evaluate the efficacy of arbidol in COVID-19 patients. A total of 100 COVID-19 patients older than 18 years were assigned to the intervention (n = 50) and control (n = 50) groups. The study exclusion criteria were respiratory, renal, or hepatic failure, anemia, thrombocytopenia, coagulation disorders, congenital heart failure, arrhythmia, and immunosuppressant therapy within 3 months before intervention. Patients in the intervention group received hydroxychloroquine (400 mg on day 1), followed by lopinavir/ritonavir (400 mg, twice daily). In comparison, the control group received hydroxychloroquine (400 mg, twice daily on day 1), followed by arbidol (200 mg, three times daily) for 7–14 days. The mean age of patients in the arbidol and lopinavir/ritonavir groups was 56.6 and 56.2 years, respectively. A majority of the patients were male (66% in the arbidol group and 54% in lopinavir/ritonavir group). As the primary outcomes, the duration of hospitalization and clinical improvement were evaluated after 7 days of treatment. Clinical improvement was defined as recovery from cough, dyspnea, and fever. Moreover, oxygen saturation, laboratory parameters, death rate, changes in CT scan after 30 days of treatment, and need for mechanical ventilation during the study were evaluated as secondary outcomes. The duration of hospitalization in the arbidol group was significantly shorter than in the lopinavir/ritonavir group (7.2 days vs. 9.6 days, p = 0.02). The oxygen saturation was significantly improved in the arbidol group compared to the lopinavir/ritonavir group (93.9% vs. 92%, p = 0.02). Of note, patients with diabetes had a longer duration of hospitalization in this study. No major adverse effects of drugs were observed; the only adverse effect was nausea and vomiting (33% in arbidol group vs. 58% in lopinavir/ritonavir group). The white blood cell (WBC) count and erythrocyte sedimentation rate (ESR) were significantly higher in the lopinavir/ritonavir group after 7 days of treatment. Overall, patients in the arbidol group had a better laboratory and clinical outcomes than the lopinavir/ritonavir group [84]. The strengths of this trial were evaluation of clinical outcomes in patients with different comorbidities and classification of patients as having mild, moderate, and severe forms of the disease based on chest X-ray and CT scan.
A single-center, retrospective clinical trial was carried out to assess the efficacy of arbidol in preventing COVID-19 in health professionals in China. A total of 164 individuals (63 male and 101 female) with a median age of 37 years were enrolled. Patients were assigned to infected (n = 82) and uninfected (control) (n = 82) groups. A prophylactic dose of oral arbidol (200 mg once daily) was administered to 23.2% (n = 19) of infected and 58.5% (n = 48) of uninfected patients. There was a significant correlation between SARS-CoV-2 infection and a prophylactic dose of arbidol (23.2% vs. 58.5%; OR, 0.214; 95% CI, 0.109-0.420; p < 0.001), which indicates the promising role of arbidol in preventing COVID-19 [121]. However, there was no significant difference in hospitalization rate and the duration of positivity of throat swab tests. Among the patients who received prophylactic doses of arbidol, the rate of infection was significantly lower (p <0.001). It was concluded that a prophylactic dose of arbidol might be a promising option for high-risk populations such as health care professionals. This study had some limitations; it was not a double-blinded study, and it was conducted at a single-center.
A retrospective cohort study was carried out in two different centers to assess the effectiveness of arbidol in COVID-19 patients with pneumonia. Of 111 patients, 49 were assigned to receive arbidol (200 mg three times daily) plus empirical treatment, and 62 were assigned to receive only empirical treatment, including INF-alpha, lopinavir/ritonavir, favipiravir, darunavir/cobicistat, and ribavirin. There was no significant difference in age, sex, or comorbidities between the two groups (p > 0.05). Virologic conversion, receiving oxygen therapy (high-flow nasal catheter), and chest CT image were evaluated as the clinical outcomes. The results showed that arbidol accelerates the viral clearance process. The rate of stable virologic conversion was 46.9% in the arbidol group and 30.6% in the control group (p = 0.079). The rate of improvement in radiologic findings was higher in the arbidol group with 55.1% vs. 32.2% in the control group (p = 0.016). Moreover, oxygen requirement was significantly reduced in the arbidol group compared to the control group (6.1% vs. 29%; p = 0.002). This study showed that clinical improvement due to arbidol therapy depends on disease severity. Patients with mild COVID-19 benefit more from arbidol treatment than patients with severe COVID-19 [117]. The only reported side effect of arbidol was bradycardia in one patient. The limitations of this trial were the small sample size and the fact that patients had not completed their treatment by the end of the intervention.
A retrospective study was carried out on 504 hospitalized patients with COVID-19 in China to assess the efficacy of arbidol hydrochloride in reducing mortality. The mean age ± SD of the patients was 59.5 ± 14.9 years, and 52% of the patients had comorbidities. All of the patients received antiviral medications. Adjustments were made based on age, sex, comorbidities, oxygen saturation, and lesion size. Arbidol was administered to 257 patients (51%). As clinical outcomes, mortality rate and lesion size (based on chest CT scan) were evaluated. This cohort study showed a reduction in the mortality rate in hospitalized patients treated with arbidol (7% vs. 24.7%; OR, 0.330; 95% CI, 0.124–0.411) [70]. After adjustment of antiviral medications, the odds ratio for reduction of the mortality rate in the arbidol group was 0.183; 95% CI, 0.075–0.446; p < 0.001. Moreover, the average decrease in lesion size was 46.43 ± 29% in the arbidol group versus 36.8 ± 24.95% in the non-arbidol group. Given the fact that this trial did not consider oxygen therapy in clinical outcomes, and because of sampling bias, the results of this trial could not be generalized [70].
A retrospective study including 50 patients with reverse transcription polymerase chain reaction (RT-PCR)-confirmed COVID-19 was conducted to compare the effectiveness of arbidol and lopinavir/ritonavir. Patients were assigned to the arbidol group (n = 16) with a median age of 26.5 (23.3–52.5) or the lopinavir/ritonavir group (n = 34) with a median age of 40.5 (34.8–52.3). The dose of arbidol was 200 mg, three times a day, and the dose of lopinavir/ritonavir was 400/100 mg, twice daily for a week. Based on data analysis, the patients treated with arbidol had better clinical outcomes on day 14 after admission than those who received lopinavir/ritonavir. The viral load in the arbidol group was undetectable on day 14 after admission, whereas, 44.1% of the patients in the lopinavir/ritonavir group had a detectable viral load [128]. No adverse effects were reported in this trial. Although this study showed beneficial effects of arbidol in viral clearance, a limitation of the study was that the clinical outcome of the patients was not reported.
A prospective, multicenter RCT was carried out in China to compare the safety and efficacy of favipiravir and arbidol in COVID-19 patients. A total of 236 patients with confirmed COVID-19 (based on Chinese guidelines) were randomly divided into two groups: 120 in the arbidol group and 116 in the favipiravir group. All of the patients received conventional Chinese medications. Patients in the arbidol group received 200 mg of arbidol three times a day for 10 days, and patients in the favipiravir group received 1600 mg twice daily on the first day and 600 mg twice daily for the remaining 9 days. The recovery rate within 7 days of treatment was evaluated as the primary outcome. Recovery was evaluated based on the following criteria for more than 72 h: axillary temperature ≤ 36.6 °C, RR ≤ 24/min, oxygen saturation ≥ 98% without supplemental oxygen therapy, relief of cough or presence of mild cough. A larger number of patients with severe illness were observed in the favipiravir group than in the arbidol group. However, after 7 days, no difference was observed in the recovery rate of the patients in the favipiravir and arbidol groups (61.21% and 51.67%, respectively; difference in recovery rate, 0.0954; 95% CI, 0.0305–0.2213; p = 0.1396) [21]. Patients’ eligibility for enrollment in this trial could be considered a limitation, since the patients were diagnosed with COVID-19 based on Chinese guidelines rather than an RT-PCR test.
Despite its potential benefit against COVID-19 in clinical trials, several retrospective studies have failed to confirm the beneficial effects of arbidol. This may have been due to limitations such as small sample size or inadequate study design [25, 56, 61, 68, 69, 118].
According to clinicaltrials.gov, two ongoing RCTs are being carried out in Iran and China to evaluate the effectiveness of arbidol in COVID-19 patients.
A systematic review and meta-analysis of 12 studies with a total of 1052 COVID-19 patients was done by Huang et al. to assess the effectiveness and safety of arbidol for COVID-19 treatment. All 12 studies (ten retrospective studies and two RCTs) took place in China, with minimum and maximum sample sizes of 32 and 236, respectively. The main outcomes used to evaluate the effectiveness of arbidol were a negative PCR result within 7 and 14 days of treatment as a primary outcome and relief of fever and cough within 7 days of treatment as a secondary outcome. The data analysis showed that administration of arbidol had a significant correlation with negative RT-PCR results within 14 days after treatment (RR, 1.27; 95% CI, 1.04–1.55; I2, 63%). However, there was no correlation between administration of arbidol and negative RT-PCR results within 7 days after treatment (RR, 1.09; 95% CI, 0.91–1.31; I2, 44%). Moreover, no correlation was observed between arbidol and relief of fever and cough as secondary outcomes (RR, 1; 95% CI, 0.91–1.1; I2, 0% and RR, 1; 95% CI, 0.85–1.18; I2, 0%, respectively). Arbidol was considered safe in this study (RR, 1.29; 95% CI, 0.57–2.92; I2, 56%) [55].
Overall, arbidol is well tolerated agent. The only adverse effect was diarrhea, reported in 10% of the patients enrolled in one clinical trial [121]. Moreover, a short duration of fever (less than 7 days) with arbidol was reported in a retrospective study [128].
VIR-2703 (ALN-COV)
VIR-2703, an inhaled agent, is a likely candidate for the prevention and treatment of COVID-19. VIR-2703 is an investigational agent that functions by an RNA interference (RNAi) mechanism in which small interfering RNA (siRNA) causes gene silencing. There are two types of RNAi targets against COVID-19: viral proteins that play a role in viral replication, such as 3CL-protease and RdRp, and host proteins that are involved in viral entry or trafficking of the virus in the host cell, such as ACE2 [107]. VIR-2703 targets conserved regions in the SARS-CoV-2 genome to inhibit viral replication [3, 107]. Moreover, VIR-2703 can also be used to control COVID-19-related cytokine release syndrome [107]. At present, the safety of the siRNA remains unclear; however, VIR-2703 is delivered by inhalation to minimize adverse effects [107]. Akerstrom et al. expressed a specifically designed siRNA in Vero E6 cells. This siRNA was designed to silence specific SARS-CoV proteins, including the 7a/7b, 3a/3b, and S proteins. The study results showed that the 3a and 7a proteins play pivotal roles in the replication of SARS-CoV [3]. In studies using a live-virus model, VIR-2703 significantly inhibited SARS-CoV-2 with EC50 <100 pM and EC95 <1 nM. Some manufactures are planning to apply to the FDA for starting a clinical trial.
Alpha-1 antitrypsin (A1AT)
Alpha-1 antitrypsin (A1AT) (also called alpha-1 proteinase inhibitor) is a glycoprotein that is produced naturally in the liver. It has been approved by the FDA for managing A1AT deficiency in patients with emphysema, at a dose of 60 mg/kg as weekly infusions [111, 122]. A1AT deficiency is correlated with an increased risk of emphysema and bronchiectasis [33, 120]. Also, A1AT has been used to treat influenza A and B [51]. This agent has antiviral and anti-inflammatory properties and is able to inhibit IL-6, IL-8, TNF-alpha, and neutrophil elastase [33, 122]. Furthermore, A1AT is a TMPRSS2 inhibitor that plays a role in the entry of SARS-CoV-2 into the host cell [121]. Although, intravenous (IV) infusion of A1AT appears to be safe and well tolerated, some adverse effects such as flu-like symptoms, hypersensitivity, the transmission of infectious agents, and potential volume overload may occur. A1AT is contraindicated in patients with immunoglobulin A (IgA) deficiency with antibodies against IgA [36]. In vitro studies showed an inhibitory effect of A1AT against SARS-CoV-2 in Caco2 and Vero cells [33].
Three ongoing clinical trials are being carried out to evaluate the effectiveness of A1AT in COVID-19 patients. These trials are RCTs based in Spain, the United States, and Saudi Arabia. The study sample sizes range from 100 to 150 patients with a total sample size of 350. Two studies were designed to enroll hospitalized COVID-19 patients to assess the effect of 120 mg/kg of A1AT administered intravenously on the first and eighth day of the intervention in addition to standard-of-care medications. In addition, inhaled A1AT will be evaluated in COVID-19 patients at a dose of 8 mL twice daily for 5 days. The mortality rate and requirement for mechanical ventilation or ICU admission will be measured in hospitalized COVID-19 patients as primary outcomes within 15 days after initiation of treatment.
Bemcentinib
Bemcentinib inhibits AXL kinase, a protein with two mechanisms of action: blocking viral entry by acting as a co-receptor for ACE2 and enhancing the type 1 IFN response against the virus in the host. Considering the critical role of AXL overexpression in the development of aggressive metastatic cancers through drug resistance and immune escape, numerous clinical trials are being carried out to evaluate the efficacy of bemcentinib in hematological cancer and solid-tumor treatment protocols. Bemcentinib plays a crucial role in the treatment of various cancers by preventing immune evasion, drug resistance, and metastasis [6, 28]. Studies have been done on Vero E6 and human ACE2-expressing A549 lung cancer cell lines to evaluate the in vitro efficacy of bemcentinib. It has been demonstrated that the AXL increases the ability of SARS-CoV-2 to infect these cell lines. Bemcentinib also reduces the viral load in cells by inhibiting ACE2-mediated viral cell entry by receptor-mediated endocytosis rather than direct fusion at the cell surface. Furthermore, bemcentinib has shown promising effects in infected mice [80].
The ACCORD [Accelerating COVID-19 Research and Development) study is a multicenter RCT designed in the UK to evaluate the safety and efficacy of candidate agents in addition to the standard of care in hospitalized COVID-19 patients [114]. The candidate agents are bemcentinib, MEDI3506, acalabrutinib, zilucoplan, and nebulized heparin. This study will be carried out in two stages, in which stage 2 (with 126 individuals for each candidate) will be adjusted based on the results of stage 1 (with 60 individuals for each candidate). It is estimated that a total of 1800 individuals will be enrolled. The treatment protocol includes 400 mg (4 capsules) of bemcentinib on the first 3 days, followed by 200 mg for the next 12 days.
A phase 2 multicenter RCT with 120 patients has been designed in India to evaluate the effectiveness of bemcentinib for treatment of hospitalized COVID-19 patients. In that study, 56 and 58 patients have been assigned to the bemcentinib and the control group, respectively.
Apilimod dimesylate (LAM-002A)
Apilimod dimesylate is an oral selective phosphatidylinositol-3-phosphate 5 kinase (PIKfyve) inhibitor that blocks the production of IL-12 and IL-23 and interferes with the entry and trafficking of SARS-CoV-2 in host cells [43]. Notably, apilimod is associated with severe immunosuppression in COVID-19 patients by inhibiting the protease proteins of the host cells and consequent inhibition of viral entry [11]. Clinical trials have shown apilimod to be a well-tolerated drug. Mild to moderate adverse effects have been observed with apilimod, including headache, fatigue, dizziness, and nausea [43]. Apilimod is used in the treatment of B-cell non-Hodgkin lymphoma (B-NHL) and could be used as monotherapy or combination therapy with a full dose of anti-CD20 (rituximab) or anti-programmed death-1 (PDL1) (atezolizumab) antibodies [35, 43]. Based on the satisfactory results of in vitro studies of apilimod against SARS-CoV-2 [62], a phase 2, double-blinded, placebo-controlled RCT is being carried out to evaluate the effectiveness of apilimod dimesylate for the prevention of progression of COVID-19 in out-patients. In total, 142 confirmed COVID-19 patients will be randomly assigned to the intervention and the control groups at a 1:1 ratio. Patients in the intervention group will receive 125 mg apilimod dimesylate orally, twice daily for 10 days. Likewise, patients in the control group will receive five capsules of a placebo, twice daily for 10 days. All of the patients will receive standard-of-care therapy. The primary outcome of this study will be changes in viral load, measured by RT-PCR. Safety and tolerability of apilimod, alteration in clinical status of patients, and clinical efficacy of apilimod will be evaluated in this study as secondary outcomes.
ONO-5334
ONO-5334 is a cysteinyl cathepsin K inhibitor that inhibits SARS-CoV-2 replication in infected lung cells [93, 94]. ONO-5334 is a drug candidate for the treatment of osteoporosis; however, the designed phase 2 clinical trial has been discontinued. ONO-5334 is tolerable and safe up to a dose of 300 mg/day for a maximum of 12 months of treatment [93, 94]. In a study by Riva et al., ONO-5334 showed antiviral effects against SARS-CoV-2 in various cell lines, including Vero E6 (EC = 0.41 µM), HEK293T (EC50 = 0.042 µM), and Huh-7 cells (EC50 = 0.078 µM), as well as primary human cells [93, 94]. ONO-5334 reduced the viral load by 72% in primary human cells (p = 0.0171) [93, 94].
MDL-28170
MDL-28170 is an inhibitor of cysteinyl cathepsin B and calpain 3 [66]. It blocks SARS-CoV-2 entry into human cells by inhibiting proteolytic processing of the viral S protein. In vivo studies have demonstrated the neuroprotective ability of MDL-28170 in a rat model with global brain ischemia in which it was found that MDL-28170 is readily absorbed through the blood-brain barrier (BBB) [66]. The in vitro antiviral effect of MDL-28170 against SARS-CoV-2 has been studied in Vero E6, Huh-7, and HEK293T cells, and the reported EC50 values were 0.22, 0.086, and 0.021 µM, respectively [93, 94]. MDL-28170 reduced the viral load by 65% in primary human cells (p < 0.0001) [93, 94].
Investigational antivirals with an inhibitory effect on the viral RdRp
Molnupiravir (MK-4482, EIDD-2801)
Molnupiravir (MK-4482; Merck) is a pro-drug of the nucleoside analogue N4-hydroxycytidine (NHC) with antiviral activity against a broad range of RNA viruses, including SARS-CoV, influenza virus, Ebola virus, and Venezuelan equine encephalitis virus (VEEV) [30, 100]. Molnupiravir is an RdRp inhibitor that binds to the enzyme’s active site [101, 103]. Moreover, it suppresses spread of the virus [30]. It is believed that molnupiravir increases the viral mutation rate beyond a biologically bearable threshold, leading to viral extinction [85]. The antiviral mechanism of molnupiravir was first demonstrated with Venezuelan equine encephalitis virus (VEEV) to occur through the accumulation of destructive mutations in the viral RNA [100]. Molnupiravir was developed to treat HCV infections in the early 2000s [100], and it has also been used in the treatment of influenza. The antiviral activity of NHC was assessed in human lung epithelial (Calu 3) cells infected with Middle East respiratory syndrome coronavirus (MERS-CoV). Virus replication in the cultures was measured for 48 h in this in vitro study. The study showed a dose-dependent reduction in viral load (half-maximal inhibitory concentration [IC50] = 0.08 µM) with no toxicity observed with the selected doses of the drug [100]. The antiviral activity of molnupiravir was also studied in Vero cells. Data analysis indicated that NHC is an effective antiviral against SARS-CoV (IC50 = 0.3 µM) [100]. Also, in human airway epithelial (HAE) cell culture, a similar dose-dependent reduction with molnupiravir was observed (> 3 logs at 10 µM) without any toxicity [100]. Cox et al. investigated the efficacy of molnupiravir in a ferret model, which simulated the spread of the virus in the human population, since both transmit the virus with mild clinical signs [30]. This study showed that molnupiravir taken twice daily could significantly reduce the viral load in the respiratory tract and suppress the chain of virus transmission [30]. Rosenke et al. evaluated the effect of MK-4482 in a Syrian hamster model [96]. Another study revealed that MK-4482 significantly decreases both the replication of SARS-CoV-2 and pathologic changes in lung cells [100]. Merck has designed an in vivo study to assess the safety profile of molnupiravir given at higher doses and for a longer duration than in previous clinical trials; this study did not find a mutagenic effect of molnupiravir in mammals.
Painter et al. designed a phase 1 RCT to evaluate the safety, tolerability, and pharmacokinetics of a single dose as well as increasing oral doses of molnupiravir in healthy individuals. A total of 130 eligible individuals were randomly assigned to the molnupiravir or placebo group at a 3:1 ratio. In the molnupiravir group, 64 subjects received a single increasing dose of molnupiravir (50–1600 mg), and 55 received multiple increasing doses of molnupiravir (50–800 mg, twice daily for 5.5 days). In this trial, the effect of food on molnupiravir was also evaluated, with 10 patients receiving 200 mg of molnupiravir in the fed state and 200 mg of molnupiravir in the fasted state after a 14-day wash-out period. The patients’ ages ranged from 19 to 60 (the majority were male) with similar demographic characteristics. Adverse effects were reported in 37.5% and 44.6% of participants in the single- and multiple-increasing-dose groups, respectively. The most common adverse effect in those receiving a single increasing dose was headache (moderate, grade 2) with a prevalence of 18.8% and 12.5% in the placebo and molnupiravir group, respectively. No grade 3 adverse effects were reported in the single-increasing dose group. The most common adverse effect in those receiving a multiple increasing dose was diarrhea (mild, grade 1) with a prevalence of 7.1% in both the placebo and molnupiravir groups. One case of a moderate adverse effect was observed in the multiple-increasing-dose group (pain in extremities and oropharynx, flu-like symptoms). In this study group, one participant who received molnupiravir 800 mg twice daily was excluded due to a mild drug-related rash. There were no dose-related changes in laboratory parameters, vital signs, or electrocardiogram (ECG). The pharmacokinetic studies revealed that absorption rate, time to maximum plasma concentration (Tmax), and maximum serum concentration (Cmax) were lower in the fed state; however, the extent of absorption was the same in the fed and fasted states [85].
A multi-center, phase 2a trial was designed with an enrollment of 202 non-hospitalized COVID-19 patients to evaluate the safety and tolerability of molnupiravir. RT-PCR-confirmed COVID-19 patients who had the signs and symptoms of the disease within 7 days were enrolled in this trial. As the primary outcome, the duration of the positive PCR was evaluated in this study. Moreover, the duration of the positive culture of the virus was measured as a secondary outcome. Based on data analysis, a significant reduction in positive cell culture was observed within 5 days after treatment of molnupiravir compared to placebo (p = 0.001). No serious adverse effect was reported in this trial. No more data have been published from this trial so far.
At the time of writing this review, four ongoing clinical trials are being carried out to evaluate the effectiveness, safety, and pharmacokinetics of molnupiravir in COVID-19 patients. Both hospitalized (n = 304) and non-hospitalized (n = 1850) COVID-19 patients will be studied in two different multi-center trials. The treatment dosages of molnupiravir in designed trials are 200–400–800 mg, twice daily for 5 days. The study sample sizes ranged from 80 to 1450, with a cumulative sample size of 3034. As primary outcomes, adverse effects of molnupiravir, viral load, sustained recovery (defined by ready-to-be discharged or non-hospitalized patients), hospitalization rate, all-cause mortality, and the WHO scale score will be evaluated in these trials.
Galidesivir (BCX4430) (immucillin-A)
Galidesivir is a broad-spectrum antiviral drug that was initially developed for the treatment of Ebola [91]. It has an inhibitory effect against a wide range of RNA viruses, including coronaviruses, HCV, Ebola virus, and yellow fever virus [75, 110]. Galidesivir is a nucleoside that inhibits the viral RNA polymerase by causing RNA chain termination [110]. According to a molecular docking study, galidesivir is predicted to act as an inhibitor of the RdRp or SARS-CoV-2 [40]. It binds to the catalytic center of the RdRp and inhibits viral replication [75]. Galidesivir has shown inhibitory activity against MERS-CoV in Vero E6 cell culture [67, 68]. In vivo studies in mice, rats, guinea pigs, and cynomolgus macaques have demonstrated an inhibitory activity of galidesivir against SARS-CoV-2 virus [110].
A phase 1b, placebo-controlled, double-blinded, range-dosing RCT has been designed to evaluate the pharmacokinetic properties, safety, and efficacy of galidesivir in hospitalized patients in Brazil with yellow fever or COVID-19. The optimum dose of galidesivir will be assessed before the study by evaluating three cohorts (eight patients in each cohort randomized in a 3:1 ratio) with different doses. Patients in each group will receive 10 mg/kg loading dose followed by 2 mg/kg, 10 mg/kg followed by 5 mg/kg, and 20 mg/kg followed by 5 mg/kg in cohorts 1, 2, and, 3, respectively. After obtaining the optimum treatment dose, 42 patients with COVID-19 will be randomly divided in a 2:1 ratio to receive galidesivir or placebo. Patients will receive galidesivir or placebo via IV infusion twice daily for 7 days. As primary outcomes, adverse effects, changes in laboratory data, and concentration of galidesivir in plasma will be measured in this trial. Patients will be followed up within 56 days after intervention.
Investigational antivirals blocking viral replication
Emetine
Emetine is an isoquinoline alkaloid derived from ipecac root that has been approved by the FDA for the treatment of amebiasis with a dosage of 1 mg/kg/day [65]. Emetine has anti-malaria and anti-inflammatory properties. Moreover, it has shown antiviral activity against various DNA and RNA viruses such as Zika virus and Ebola virus [14, 27, 50, 108, 123]. Previous in vitro studies have demonstrated the inhibitory activity of emetine against MERS-CoV and SARS-CoV [38, 101]. The suggested mechanism of emetine is decreasing viral RNA and protein synthesis by blocking the interaction of SARS-CoV-2 RNA and eukaryotic translation initiation factor 4E (eIF4E) [65]. eIF4E plays an essential role in viral protein translation, leading to viral replication.
Emetine can also decrease the lipopolysaccharide-mediated IL-6 level and moderately reduce the level of TNF-alpha in M1-polarized THP-1 macrophages. Due to the central role of inflammation in the pathophysiology of COVID-19, emetine could be a potential therapeutic candidate. The maximum daily dose of emetine is 60 mg, with a maximum tolerated dose of 10 mg/kg [108].
An in vitro study confirmed the anti-SARS-CoV-2 activity of emetine in Vero cells, with an estimated 50% effective concentration (EC50) of 0.46 μM for reducing the titer of infectious virus and 0.5 μM for reducing the RNA copy number. This study also showed a synergistic effect of emetine and remdesivir, which could decrease the effective concentration of each compound to below the maximum beneficial plasma concentration [27]. Another in vitro study indicated a potent antiviral activity of low-dose emetine in Vero cells, with an EC50 of 0.007 μM, making it approximately 30-fold more potent than remdesivir. Also, an in vivo pharmacokinetic analysis showed high emetine concentrations in the lung tissue that were 200 times higher than the EC50 or emetine [108].
Fan et al., in a multi-center RCT, evaluated the efficacy of low-dose emetine in 63 patients with RT-PCR-confirmed COVID-19 in hospitals in Wuhan and Anhui provinces of China. Patients aged 18–80 years with symptoms of mild or moderate COVID-19 and hospitalization due to the fever (axillary temperature of 36.7 °C or more), RR more than 24 per minute, or cough were excluded. Patients in the treatment arm (n = 37) received 3.6 mg of emetine per os, three times a day for 10 days in addition to the standard antiviral therapy (arbidol 200 mg per os, three times in a day for 10 days), whereas patients in the control group (n = 26) only received the standard antiviral therapy. No significant differences were observed in baseline characteristics of patients except for age, HR, and RR in the Anhui province hospitals. Patients in the emetine group in the Anhui hospitals were significantly younger (p = 0.03) with a higher HR (p = 0.01) and RR (p < 0.01). As a primary outcome, the comparison of the time required for negative seroconversion and the rate of negative results on day 10 showed no significant differences between the two groups (p > 0.05). A comparison of axillary temperature, oxygen saturation, and RR on days 1, 3, 5, 7, and 10 after treatment showed no significant difference between the study groups except for oxygen saturation on day 1, which was significantly higher in the emetine group (97.83 ± 1.13 vs. 97.00 ± 0.85; p = 0.03). Moreover, no adverse effects were noted in the emetine group during the study [108]. Given the small sample size, lack of data, and different baseline characteristics of the patients, the results should be interpreted with caution. Also, severe cases of COVID-19 were not included in this study. Therefore, well-designed RCTs should be carried out for further evaluation of emetine under different clinical conditions. Finally, an important advantage of emetine is that high concentrations can be achieved in target tissues such as lung, kidney, and liver [108].
Plitidepsin
Plitidepsin (Aplidin; PharmaMar) (dehydrodidemnin B) is a natural agent isolated from a Caribbean marine organism (Aplidium albicans). It is an inhibitor of eukaryotic translation elongation factor 1 alpha (eEF1A), a factor responsible for the enzymatic delivery of transfer RNA (tRNA) to the ribosome in the translation process [112]. Notably, eEF1A is an essential host factor for replication of various viruses, including influenza virus, respiratory syncytial virus (RSV), and SARS-CoV [78, 112]. White et al. showed that plitidepsin could inhibit SARS-CoV-2 replication using engineered cells that expressed a mutated version of the eEF1A protein (eEF1A-A399v) [112]. Of note, Reuschl et al. demonstrated the antiviral activity of plitidepsin against the B.1.1.7 variant in human gastrointestinal and lung epithelial cell lines, a variant that was discovered recently in the UK. Generally, since there is less likelihood of mutation of human proteins than of viral proteins, using plitidepsin as a host-targeted antiviral can prevent future viral resistance. Plitidepsin may cause toxicity by altering multiple pathways, including causing cell cycle arrest, inhibiting cell growth, and inducing apoptosis. Of note, the manufacturer claims that the doses used in COVID-19 trials are well tolerated in humans. Plitidepsin has clinical approval in Australia for the treatment of multiple myeloma (MM), the suggested mechanism of which is blocking a protein that also plays a key role in the pathogenesis of SARS-CoV-2 [7, 112]. Several clinical trials have already been carried out to evaluate the effects of plitidepsin on lymphoma, MM, liposarcoma, and prostate cancer [39]. The development of this drug has been ended due to the development of competitive agents and the lack of large clinical trials [39]. Plitidepsin is administered at 5 mg/m2 every 2 weeks in patients with relapsed/refractory MM, and it can be used in combination with immunomodulators in a multidrug regimen [39].
White et al. evaluated the cytotoxic profile and antiviral properties of plitidepsin against SARS-CoV-2 under ex vivo and in vivo conditions. The antiviral effect of plitidepsin was studied in Vero E6, hACE2-HEK293T, and pneumocyte-like cells, with IC90 values of 1.76, 0.88, and 3.14 nM, respectively. Moreover, the cytotoxic concentration (CC50) was 1.99, >200, and 65.43 nM, respectively. The results of ex vivo studies in Vero E6 and hACE2-HEK293T cells revealed that plitidepsin is more potent and more toxic than remdesivir. The IC90 of remdesivir in Vero E6 and hACE2-HEK293T cells is 2.25 and 24.20 µM, respectively, and its CC50 is >20 µM and >2000 nM, respectively. The in vivo efficacy of prophylactic plitidepsin was tested in two animal models (wild-type BALB/c mice and K18-hACE2 mice) infected with SARS-CoV-2. These mice received a prophylactic dose of plitidepsin, either 0.3 mg/kg/day for 2 days (first group), or a single dose of 1 mg/kg (second group). The mice were infected with 1 × 104 pfu of SARS-CoV-2 2 h after the dose of plitidepsin. The results showed a 2-log reduction in the respiratory viral load in the first group and a 1.5-log reduction in the second group. The toxicity of plitidepsin was proposed to be due to body weight loss in the first group of mice [112]. Moreover, plitidepsin alleviated lung inflammation in mice at 3 days postinfection compared to the control mice or the mice treated with remdesivir [112].
PharmaMar is a proof-of-concept, multi-center, randomized trial was designed to evaluate the safety, toxicity, and recommended dose of plitidepsin in hospitalized patients in Spain with RT-PCR-confirmed COVID-19. A total number of 46 patients were divided into three groups receiving plitidepsin 1.5, 2, and 2.5 mg per day, which was administered by IV infusion over 1.5 h for 3 days. The primary outcomes of the study included neutropenia, thrombocytopenia, anemia, lymphopenia, elevation of alanine transaminase (ALT) or aspartate transaminase (AST), neurotoxicity, QT prolongation, and adverse effects > grade 3. This study revealed a viral load reduction in patients between days 4 and 7 after initiation of the intervention. A viral load reduction of 50% and 70% was documented on days 7 and 15, respectively. Finally, 38.2% and 80.7% of patients were discharged within 7 and 15 days after hospitalization, respectively.
An ongoing multi-center, open-label, phase 3 RCT is being carried out in Spain to assess the safety and efficacy of plitidepsin in moderate COVID-19 patients, which is defined by the requirement for hospitalization and oxygen therapy. A total of 609 patients were randomly divided in a 1:1:1 ratio to receive 1.5 mg plitidepsin, 2.5 mg plitidepsin, or a placebo. Patients will receive IV plitidepsin in combination with 8 mg of IV dexamethasone per day, for 3 days, followed by 6 mg/day IV or oral for next 7 days. Patients in the control group will receive dexamethasone with the same dosages in addition to 5 mL of remdesivir (100 mg/20 mL vial). As the primary outcome, clinical recovery will be evaluated in patients within 31 days. Clinical recovery is defined as reaching to the WHO scale of 0–2, no re-hospitalization within 31 days after the intervention, or Barthel index > 90/100 at the time of discharge.
Trabedersen (OT-101/AP 12009)
Trabedersen (OT-101/AP 12009) is an antisense oligonucleotide that blocks the mRNA of human transforming growth factor beta 2 (TGF-β2), a protein that is required for viral replication. Xiong et al. showed that high TGF-β levels in patients with COVID-19 could lead to ARDS and Kawasaki syndrome [105, 106, 116]. Considering the pivotal role of TGF-β in the immunopathology of COVID-19, trabedersen seems to be helpful in SARS-CoV-2-related immune dysregulation [105]. Furthermore, trabedersen is a candidate adjuvant for a second generation of COVID-19 vaccines for stimulating an immune response. Most of the adverse effects observed during the treatment with trabedersen have been attributed to underlying diseases in the patients, except thrombocytopenia, which might be related to trabedersen [18]. Other related or possibly related adverse effects of the drug include a rise in ALT and gamma-glutamyl transferase (GGT) levels, decreased hemoglobin, maculopapular rash, gastrointestinal hemorrhage, fatigue, nausea, headache, vomiting, dyspnea, pyrexia, asthenia, constipation, decreased appetite, diarrhea, general physical health deterioration, pain in extremities, pruritus, chills, metastasis, and skin inflammation [18]. Trabedersen is an antineoplastic agent, since overexpression of TGF-β is associated with proliferation, metastasis, angiogenesis, and immunosuppression of tumors [57]. Potential benefits of trabedersen have been observed in patients with pancreatic cancer, glioblastoma, melanoma, and high-grade glioma. In addition, D’Cruz et al. reported a synergistic effect of trabedersen with chemotherapy in treatment of tumors [32]. The Ministry of Food and Drug Safety of Korea has approved the combination of OT101/IL-2 (trabedersen/I-2) for the treatment of patients with solid cancer. The FDA has also approved OT101 for diffuse intrinsic pontine glioma (DIPG).
A phase 2, double-blinded, placebo-controlled RCT is being carried out in Brazil to evaluate the efficacy and safety of trabedersen in addition to the standard of care in hospitalized patients with severe COVID-19. Eighteen participants with PCR-confirmed COVID-19 will be randomly assigned to the intervention (trabedersen) or placebo group in a 2:1 ratio. The regimen of artemisinin will be 140 mg/m2/day, IV infusion for 7 days. Severe COVID-19 is defined as criteria 5 or 6 in the ordinal scale of criteria of the WHO. The clinical improvement score will be measured as a primary outcome in this study within 7, 21, and 28 days after starting the drug.
AT-527 (RO7496998)
AT-527 is an oral guanosine nucleotide prodrug with direct activity against SARS-CoV-2. AT-527 is a possible candidate for use in pre- and post-exposure prophylaxis of COVID-19. The suggested mechanism for At-527 is inhibiting SARS-CoV-2 replication by blocking viral RNA polymerase [48].
A clinical trial including patients with HCV infection showed that 550 mg of AT-527 once daily for 7 days is highly effective and well tolerated [46, 47]. Based on an in vitro study, AT-511 (the free base of AT-527) potentially inhibits SARS-CoV-2 in human airway epithelial (Huh-7) cells [48], and no cytotoxicity was observed at concentrations up to 100 µM. Moreover, in a study of SARS-CoV-2-infected Vero 76 cells, the average EC90 was 0.55 µM [46, 47].
Two ongoing phase 2 RCTs are being carried out in the United States to assess the effectiveness of AT-527 in COVID-19 patients. In both studies, the dosage of AT-527 in the treatment arm is 550 mg, twice daily. Changes in viral load, adverse effects of the drug, time to relief of the symptoms, and percentage of respiratory insufficiency will be evaluated in these trials as primary outcomes.
Nitazoxanide (NTZ)
Nitazoxanide (NT-300; Romark Laboratories) is a low-cost and well-tolerated FDA-approved antiprotozoal drug with potential effects against COVID-19 [72, 95]. Nitazoxanide has shown a broad-spectrum in vitro antiviral effect against several viruses, including coronaviruses, influenza viruses, and hepatitis B and C viruses [95]. Nitazoxanide is a pro-drug that is rapidly deacetylated to its active form, tizoxanide [2-hydroxy-N-(5-nitro-2thiazolyl) benzamide] (TIZ) [72]. Nitazoxanide has dose-dependent therapeutic efficacy due to its low aqueous solubility and high permeability [72]. It has an oral bioavailability about 30%, which can increase to 50% when taken with food [72]. The FDA has approved nitazoxanide for the treatment of infectious diarrhea and enteritis caused by parasites [79]. A high dose of nitazoxanide (1 g twice daily) is indicated for AIDS-related diarrhea [72]. Moreover, nitazoxanide is approved for the treatment of diarrhea due to infection with Cryptosporidium or Giardia protozoa in immunocompromised adults and children [72]. Some clinical trials have suggested the promise of nitazoxanide for treatment of gastroenteritis, hepatitis, and influenza [72]. The dosage of nitazoxanide in intestinal parasitosis is 500 mg, twice daily for 3 days. The suggested antiviral mechanism of nitazoxanide is depletion of ATP-sensitive intracellular Ca2+ stores, which leads to phosphorylation of protein kinase R (PKR) and eukaryotic translation initiation factor 2 (eIF2), downregulation of cellular translation, and blocking of viral spread and reproduction [79]. It has been shown that nitazoxanide can balance the pro-inflammatory and anti-inflammatory responses of the host and suppresses the COVID-19-induced cytokine storm [72, 79]. Furthermore, nitazoxanide can potentiate innate host immunity and antiviral mechanisms against this virus, suggesting a lower probability of developing antiviral resistance against the drug [72]. Nitazoxanide hampers replication of SARS-CoV-2 at low concentrations in Vero CCL81 cells [95]. Moreover, it has been found to significantly reduce the viral load in Vero E6 cells (ATCC-1586) 48 h after infection [72, 95]. The effect of nitazoxanide in lowering the viral load has also been confirmed in SARS-CoV-2-infected HEK 293T and Calu-3 cells, without a loss in cell viability [95]. A physiologically-based pharmacokinetic (PBPK) modeling study has indicated that the proper doses of nitazoxanide are 1200 mg QID, 1600 mg TDS, 2900 mg BID in the fasted state and 700 mg QID, 900 mg TDS, and 1400 mg BID in the fed state to achieve effective plasma and lung concentrations against SARS-CoV-2 [72]. The maximum tolerable dose of nitazoxanide is 4 g daily [72].
Several clinical trials have been designed to evaluate the safety and effectiveness of nitazoxanide, either alone or in combination with other agents, such as hydroxychloroquine, ivermectin, or ribavirin. Moreover, combination therapy with nitazoxanide and azithromycin has been proposed [63].
A phase 2, multicenter, double-blind, placebo-controlled RCT was designed to assess the efficacy of early administration of nitazoxanide in 392 patients with mild COVID-19. Patients with dry cough, fever, and/or fatigue within 3 days of onset were selected randomly in a 1:1 ratio to receive 500 mg of nitazoxanide three times a day for 5 days or placebo. Relief of symptoms was evaluated in this trial as the primary outcome. Moreover, viral load, laboratory parameters, serum biomarkers, possible adverse effects, and hospitalization rate were evaluated as secondary outcomes. The results showed no significant difference regarding symptom relief in patients with mild COVID-19, while the viral load was lower in the intervention group than in the placebo group (p = 0.006). There was no significant difference in other outcomes between the two groups. Moreover, no serious adverse effects were observed in this trial [95]. The limitations of this study were short-term follow-up and including only a few symptoms for screening of COVID-19.
At the time of writing of this manuscript, 13 ongoing RCTs were being carried out to evaluate nitazoxanide as a treatment or for prevention of COVID-19. Six of these trials evaluate a combination of nitazoxanide with various drugs, such as ivermectin, hydroxychloroquine, atazanavir/ritonavir, ribavirin, and vitamin B-complex. Ongoing clinical trials include patients with mild to severe COVID-19. Moreover, prophylactic nitazoxanide will be studied pre- and post-exposure in healthcare workers or high-risk groups. The sample sizes of the studies range from 40 to 1407, with a total sample size of 4996. The dose of nitazoxanide varies in different trials (300, 500, 600, 1000 mg twice daily, 500 and 600 mg three times a day, and 500 mg four times a day).
Brequinar (BQR)
Brequinar is an antimetabolite that non-competitively and reversibly inhibits dihydroorotate dehydrogenase (DHODH) [88]. DHODH converts dihydroorotate to orotate in the pathway of pyrimidine biosynthesis, which is necessary for the production of nucleotides for RNA and DNA synthesis [16, 88]. Initially, brequinar was developed as an anti-neoplastic agent [7]. In vivo studies have shown that brequinar and other DHODH inhibitors are effective in the treatment of cancer and auto-immune diseases [73]. Moreover, it is used in transplantation procedures (mostly in combination with cyclosporine) to prevent and manage graft rejection [86]. It also has immunosuppressive activity because it interferes with lymphoid cell proliferation [76]. Likely side effects of brequinar include bone marrow suppression, leukocyte reduction, thrombocytopenia, and gastrointestinal disorders due to the antimetabolite activity of the drug [86]. Brequinar has a mutation-resistant antiviral effect against various RNA viruses, including SARS-CoV-2, influenza A virus, Zika virus, and Ebola virus [115, 119].
Based on the promising results obtained with brequinar against SARS-CoV-2 in Vero E6 cells, two RCTs have been designed so far [115]. In both trials, patients are given 100 mg of brequinar capsules for 5 days. The CRISIS study is a phase 1, multi-center, open-label RCT on 23 hospitalized patients with COVID-19. The second study, CRISIS2, is a phase 2, multi-center, placebo-controlled RCT on 115 outpatients with COVID-19. The safety and tolerability of brequinar will also be evaluated in these studies.
Investigational antivirals with immunological activity
Brilacidin (PMX-30063)
Brilacidin received a fast-track designation by the FDA in January 2021 due to its potential for treatment of COVID-19. It has been approved for starting phase 2 clinical trials for the treatment of hospitalized COVID-19 patients. In addition to promising results in pre-clinical studies, brilacidin has been predicted to be active against SARS-CoV-2 in in silico studies [20, 77]. It has already been tested in a phase 2 clinical trial for the treatment of acute bacterial skin and skin structure infections (ABSSSI) and ulcerative proctitis. Brilacidin is a synthetic, nonpeptidic, host defense protein/peptide (HDP) mimetic with antiviral, anti-inflammatory, and antibacterial activity. HDPs, which are components of the innate immune response, are integral proteins that interact specifically with pathogens and facilitate their proteolysis and degradation [10, 98]. Suppression of defensins and human cathelicidin (as HDPs) has been observed in cells infected with SARS-CoV-2 [31, 59]. However, in vitro studies have suggested that brilacidin interferes with the virus prior to infection, disrupting its structure and blocking entry [10]. Pre-clinical and clinical studies indicate that brilacidin inhibits the activity of IL-6, IL-1β, TNF-alpha, and other pro-inflammatory cytokines and chemokines, which are strongly associated with poor prognosis in hospitalized patients with COVID-19.
Bakovic et al. evaluated the activity of brilacidin in Calu-3 and Vero cells [10] and found that it has a potent, dose-dependent inhibitory effect on multiple strains of SARS-CoV-2. Moreover, a synergistic effect of brilacidin in combination with remdesivir was observed. The reported selectivity index for brilacidin was 426, which suggests its clinical applicability [10].
A phase 2, multi-center, double-blinded, placebo-controlled RCT is being carried out to evaluate the efficacy and safety of brilacidin in 120 hospitalized patients with moderate to severe COVID-19. Moderate COVID-19 is defined as oxygen saturation greater than 93% or RR between 20 and 30 per minute. Severe COVID-19 is defined as oxygen saturation ≤93%, oxygenation ratio <300 mmHg, or RR ≥30 per minute. Patients in the intervention arm will receive brilacidin IV for 3–5 days in addition to the standard of care. The primary outcome of this study will be the day of clinical recovery based on the ordinal scale.
Rintatolimod (ampligen)
Rintatolimod is an antiviral drug with immunomodulatory properties that is predicted to play a significant role in the treatment of acute COVID-19 and the management of COVID-19-related chronic fatigue-like symptoms. It has been estimated that 27% of patients experience chronic fatigue syndrome following COVID-19; therefore, early treatment of COVID-19 patients seems to be critical for improving clinical outcomes [19]. Rintatolimod was clinically approved in 2016 in Argentina as the first therapy worldwide for chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). However, the FDA has not yet approved rintatolimod for this purpose. Rintatolimod has been found to improve cognition, exercise tolerance, and neuropsychological health and decrease RNase L activity in people with ME/CFS [82]. Moreover, it is a possible treatment for HIV/AIDS, some types of cancer, and influenza (H1N1) [17, 41, 58]. Rintatolimod is a mismatched double-stranded RNA (dsRNA) that mimics the virus, inducing innate immunity via Toll-like receptor 3 (TLR3) agonist activity [49]. It stimulates the production of IFNs and activates IFN-induced proteins that require dsRNA for activity, such as protein kinase R [29, 49]. Rintatolimod is a well-tolerated drug. A clinical trial showed that the main adverse effect of rintatolimod is mild flu-like symptoms (13.9%), which occur during the first weeks of infusion [82]. Other adverse effects are headache, chills, fever, vasodilation, pain, injection site reaction, pruritus, diarrhea, syncope, ear disorder, nausea, and migraine [82]. According to a clinical trial carried out by Strayer et al., the well-tolerated dosage of rintatolimod is 400 mg IV twice daily [102].
A phase 1/2 clinical trial with 64 patients is being carried out in the United States in order to evaluate the efficacy and safety of rintatolimod in combination with INF alpha-2b as an effective prophylactic treatment for cancer patients with mild-to-moderate COVID-19, which is defined by confirmed COVID-19 by RT-PCR, fever or cough, shortness of breath, and nasal congestion. Patients with leukemia and allogenic hematopoietic stem cell transplantation (HSCT) will not be included in this study. Patients will receive IV rintatolimod over 2.5–3 h and IV recombinant INF alfa-2b over 20 min. Both medications will only be administered on days 1 and 3 (or 4). As primary outcomes, probable adverse effects and kinetics of viral clearance assessed by RT-PCR will be evaluated within 30 days of treatment.
Other investigational antivirals
Stannous protoporphyrin (SnPP; RBT-9; renibus therapeutics)
Stannous protoporphyrin (SnPP) is a cyclic tetrapyrrole derivative with antiviral and immune-modulating properties. The FDA has granted a fast-track designation to SnPP for COVID-19 treatment. SnPP has in vitro antiviral activity against yellow fever virus, dengue virus, Zika virus, chikungunya virus, and other enveloped viruses [83]. SnPP is a heme oxygenase 1 inhibitor [5]. In the pathophysiology of COVID-19, four proteins are potential heme binders, including the host cell proteins ACE2 and TMPRSS2 and the viral proteins 7a and S [54]. SARS-CoV-2 virus attacks the heme part of hemoglobin (a porphyrin with iron), which causes increased levels of porphyrin in the blood and hemolytic disorders in COVID-19 patients [12, 24, 54]. Accordingly, laboratory examination of 99 patients with COVID-19 pneumonia revealed increased serum ferritin level (63%) and increased ESR (85%) [21, 24, 25].
A phase 1, non-randomized clinical trial with 56 participants has been designed to evaluate the efficacy of sunlight-activated porphyrin in COVID-19 patients. The intervention group will receive 5, 7, or 9 mg of porphyrin plus an hour of sunlight exposure for 14 days.
Antroquinonol (Hocena)
Antroquinonol is a novel molecule with antiviral, anti-inflammatory, anti-fibrotic, anti-cancer, and anti-hyperlipidemia properties that was discovered in 2006 and was isolated from a mushroom used in traditional medicine, Antrodia camphorate [53]. Antroquinonol is an orphan drug designated for the treatment of pancreatic cancer, acute myeloid leukemia (AML), and hepatocellular carcinoma (HCC) [26, 124]. The anti-cancer mechanism of antroquinonol probably involves the inhibition of isoprenyltranferase, an enzyme that is involved in post-translational processing of Ras and Rho proteins [53]. The suggested mechanism of antroquinonol in HCC and pancreatic cancer involves inhibition of translation of the mammalian target of rapamycin (mTOR), which leads to cell apoptosis [26, 124]. The mechanism by which antroquinonol functions in the treatment of COVID-19 remains unclear [53]. [22] showed that antroquinonol treatment (40 mg/kg/day) could improve renal function and blood pressure in rats with endothelial dysfunction by modulating NADPH oxidase-4 (NOX4) activity and upregulating the nuclear factor erythroid 2-related factor 2 (Nrf-2) pathway [22].
A phase 2 RCT is being carried out in the United States to evaluate the effectiveness of antroquinonol in 174 hospitalized patients with COVID-19. Patients in the intervention group will receive a 100-mg capsule of antroquinonol twice daily for 14 days in addition to the standard of care. The percentage of patients without respiratory failure will be measured as the primary outcome. Moreover, days of hospitalization, changes in viral load by RT-PCR, and clinical improvement based on the WHO ordinal scale will be evaluated as secondary outcomes within the 28 days of intervention.
Discussion
In this review, we have summarized the investigational antivirals against COVID-19. Generally, these agents interfere with viral functions or those of host proteins, and most of the drugs described here are indicated for other conditions and are being repurposed now for their probable activity against SARS-CoV-2.
Our literature review showed that some of the potential antivirals currently under investigation show promise against COVID-19, as summarized below.
NO is an available, cheap, and well-tolerated antiviral with satisfactory evidence for its therapeutic and prophylactic effects. Patients with mild to severe COVID-19 and ARDS benefit from NO treatment. Moreover, NO reduces respiratory effort and oxygen requirement in patients receiving oxygen supplementation. It also prevents COVID-19 progression in infected patients. Methemoglobinemia and production of nitric dioxide (a byproduct of NO with a safety limit of 2 ppm) are the most common adverse effects of NO that should be considered in determination of the proper dose. Based on previous evidence, the minimum effective dose of NO in the treatment of bacterial pneumonia is 160 ppm—at dose at which no adverse effect was observed. Large numbers of trials are being carried out with NO at various dosages and in patients with different levels of severity of COVID-19.
Molnupiravir is another agent that has successfully passed a phase 1 clinical trial and is currently in phase 2/3 clinical trials. Molnupiravir has been shown to be effective in patients with COVID-19 by reducing the duration of infection. Molnupiravir prevents inter-individual viral transmission. Therefore, it might be beneficial for use in individuals for whom a vaccine is not available or who are unwilling to be vaccinated. Painter et al., in an RCT, showed that molnupiravir is safe, with a few mild or moderate adverse effects such as diarrhea and headache [85]. Due to the significant efficacy of molnupiravir treatment, EUA or full authorization of molnupiravir is expected.
Plitidepsin has shown in vivo and ex vivo antiviral effects against SARS-CoV-2 and is almost 27-fold more potent than remdesivir, the first FDA-approved drug against COVID-19. Plitidepsin prevents viral resistance, since it affects a host protein rather than a viral target.
Of the antivirals discussed here, arbidol is the agent with the most controversial results regarding COVID-19 patients. Arbidol did not demonstrate good efficacy in patients with severe COVID-19. Moreover, it has been shown that clinical outcomes of treatment with arbidol depend on disease severity. These issues necessitate more RCTs with large sample sizes to evaluate the effectiveness of arbidol in patients with COVID-19. Nojomi et al. compared the effectiveness of arbidol and lopinavir/ritonavir in a non-randomized clinical trial and showed that arbidol could significantly decrease the mortality rate [84].
Limitations
This review may have some limitations. First, due to mounting data in the COVID-19 field, we could not include all of the evidence, and we therefore focused more on completed and ongoing clinical trials. Second, due to daily publishing of data concerning investigational drugs, the importance of some agents may have changed, and some agents might have obtained FDA approval or had their approval revoked. The design of appropriate RCTs for agents with promising preclinical effects should eventually allow us to assess the best therapeutic choices for the treatment of COVID-19.
Conclusion
This review summarizes the novel investigational antivirals against COVID-19. Based on the current evidence, molnupiravir, plitidepsin, and NO are predicted to play a major role in the treatment of COVID-19 in the near future. There is an urgent need to design more RCTs to evaluate the safety and efficacy of repurposed investigational antivirals for the treatment of COVID-19.
References
Akaberi D, Krambrich J, Ling J, Luni C, Hedenstierna G, Järhult JD et al (2020) Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol 37:101734
Akerström S, Gunalan V, Keng CT, Tan YJ, Mirazimi A (2009) Dual effect of nitric oxide on SARS-CoV replication: viral RNA production and palmitoylation of the S protein are affected. Virology 395(1):1–9
Akerström S, Mirazimi A, Tan YJ (2007) Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antiviral Res 73(3):219–227
Akiyama T, Hirata T, Fujimoto T, Hatakeyama S, Yamazaki R, Nomura T (2020) The natural-mineral-based novel nanomaterial IFMC increases intravascular nitric oxide without its intake: implications for COVID-19 and beyond. Nanomaterials (Basel) 10(9):1699
Al-Horani RA, Kar S, Aliter KF (2020) Potential anti-COVID-19 therapeutics that block the early stage of the viral life cycle: structures, mechanisms, and clinical trials. Int J Mol Sci 21(15):5224
Arechederra M, Bazai SK, Abdouni A, Sequera C, Mead TJ, Richelme S et al (2021) ADAMTSL5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. J Hepatol 74(4):893–906
Arteaga CL, Brown TD, Kuhn JG, Shen HS, O’Rourke TJ, Beougher K et al (1989) Phase I clinical and pharmacokinetic trial of Brequinar sodium (DuP 785; NSC 368390). Cancer Res 49(16):4648–4653
Attia YA, El-Saadony MT, Swelum AA, Qattan SYA, Al-Qurashi AD, Asiry KA et al (2021) COVID-19: pathogenesis, advances in treatment and vaccine development and environmental impact-an updated review. Environ Sci Pollut Res Int 28(18):22241–22264
Bagate F, Tuffet S, Masi P, Perier F, Razazi K, de Prost N et al (2020) Rescue therapy with inhaled nitric oxide and almitrine in COVID-19 patients with severe acute respiratory distress syndrome. Ann Intensive Care 10(1):151
Bakovic A, Risner K, Bhalla N, Alem F, Chang TL, Weston WK et al (2021) Brilacidin demonstrates inhibition of SARS-CoV-2 in cell culture. Viruses 13(2):271
Baranov MV, Bianchi F, van den Bogaart G (2020) The PIKfyve inhibitor apilimod: a double-edged sword against COVID-19. Cells 10(1):30
Barcellini W, Fattizzo B (2015) Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers 2015:635670
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC et al (2020) Remdesivir for the treatment of Covid-19—final report. N Engl J Med 383(19):1813–1826
Bleasel MD, Peterson GM (2020) Emetine is not ipecac: considerations for its use as treatment for SARS-CoV2. Pharmaceuticals (Basel) 13(12):428
Boriskin YS, Leneva IA, Pécheur EI, Polyak SJ (2008) Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr Med Chem 15(10):997–1005
Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, Rohlena J (2020) Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim Biophys Acta Mol Basis Dis 1866(6):165759
Brodsky I, Strayer DR, Krueger LJ, Carter WA (1985) Clinical studies with ampligen (mismatched double-stranded RNA). J Biol Response Mod 4(6):669–675
Cabala K, Earabino J, Pacult P, Uckun F (2020) Rationale for a randomized, placebo-controlled, phase 2 study of Rejuveinix (RJX) in COVID-19 patients with acute lung injury and hypoxemic respiratory failure. Clin Investig (Lond) 10(2):185–189
Carfì A, Bernabei R, Landi F (2020) Persistent symptoms in patients after acute COVID-19. JAMA 324(6):603–605
Cavasotto CN, Di Filippo JI (2021) In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol Inform 40(1):e2000115
Chen C, Zhang Y, Huang J, Yin P, Cheng Z, Wu J, et al (2020) Favipiravir versus arbidol for COVID-19: a randomized clinical trial. medRxiv 2020.03.17.20037432
Chen JR, Ko J, Yeh WJ, Huang WC, Yang HY (2018) Renoprotective effects of antroquinonol in rats with N(ω)-nitro-l-arginine methyl ester-induced hypertension. Nutrients 10(10):1521
Chen L, Liu P, Gao H, Sun B, Chao D, Wang F et al (2004) Inhalation of nitric oxide in the treatment of severe acute respiratory syndrome: a rescue trial in Beijing. Clin Infect Dis 39(10):1531–1535
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395(10223):507–513
Chen X, Zhang Y, Zhu B, Zeng J, Hong W, He X, et al (2020) Associations of clinical characteristics and antiviral drugs with viral RNA clearance in patients with COVID-19 in Guangzhou, China: a retrospective cohort study. medRxiv 2020.04.09.20058941
Chiang PC, Lin SC, Pan SL, Kuo CH, Tsai IL, Kuo MT et al (2010) Antroquinonol displays anticancer potential against human hepatocellular carcinoma cells: a crucial role of AMPK and mTOR pathways. Biochem Pharmacol 79(2):162–171
Choy KT, Wong AY, Kaewpreedee P, Sia SF, Chen D, Hui KPY et al (2020) Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res 178:104786
Ciardiello D, Blauensteiner B, Matrone N, Belli V, Mohr T, Vitiello PP et al (2021) Dual inhibition of TGFβ and AXL as a novel therapy for human colorectal adenocarcinoma with mesenchymal phenotype. Med Oncol 38(3):24
Clemens MJ, Elia A (1997) The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res 17(9):503–524
Cox RM, Wolf JD, Plemper RK (2021) Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat Microbiol 6(1):11–18
Crane-Godreau MA, Clem KJ, Payne P, Fiering S (2020) Vitamin D deficiency and air pollution exacerbate COVID-19 through suppression of antiviral peptide LL37. Front Public Health 8:232
D’Cruz O, Lee C, Trieu V, Hwang L (2017) Synergistic antitumor effects of OT-101 (trabedersen), a transforming growth factor-beta 2 (TGF-β2) antisense oligonucleotide (ASO) and chemotherapy in preclinical tumor models. Ann Oncol 28:v583–v584
de Loyola MB, Dos Reis TTA, de Oliveira GXLM, da Fonseca Palmeira J, Argañaraz GA, Argañaraz ER (2021) Alpha-1-antitrypsin: a possible host protective factor against Covid-19. Rev Med Virol 31(2):e2157
de Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O’Connor R, et al (2020) Comparative study of a 3CL (pro) inhibitor and remdesivir against both major SARS-CoV-2 clades in human airway models. bioRxiv
Diefenbach C, Cohen J, Harb W, Ansell S, Nastoupil L, Abramson J et al (2020) Results of a completed phase I study of LAM-002 (apilimod dimesylate), a first-in-class phosphatidylinositol-3-phosphate 5 kinase (PIKfyve) inhibitor, administered as monotherapy or with rituximab or atezolizumab to patients with previously treated follicular lymphoma or other B-cell cancers. J Clin Oncol 38:8017
Dirksen A, Piitulainen E, Parr DG, Deng C, Wencker M, Shaker SB et al (2009) Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 33(6):1345–1353
Dunia A, Sanaa B (2020) The potential role of inhaled nitric oxide in managing COVID-19 associated lung complications review of literature. Int J Respir Pulm Med 7(4)
Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J et al (2014) Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 58(8):4885–4893
El Bairi K, Trapani D, Petrillo A, Le Page C, Zbakh H, Daniele B et al (2020) Repurposing anticancer drugs for the management of COVID-19. Eur J Cancer 141:40–61
Elfiky AA (2020) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci 253:117592
Essey RJ, McDougall BR, Robinson WE Jr (2001) Mismatched double-stranded RNA (polyI-polyC(12)U) is synergistic with multiple anti-HIV drugs and is active against drug-sensitive and drug-resistant HIV-1 in vitro. Antiviral Res 51(3):189–202
Gagarinova VM, Ignat’eva GS, Sinitskaia LV, Ivanova AM, Rodina MA, Tur’eva AV (1993) The new chemical preparation arbidol: its prophylactic efficacy during influenza epidemics. Zh Mikrobiol Epidemiol Immunobiol 5:40–43
Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P et al (2017) Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 129(13):1768–1778
Gebistorf F, Karam O, Wetterslev J, Afshari A (2016) Inhaled nitric oxide for acute respiratory distress syndrome (ARDS) in children and adults. Cochrane Database Syst Rev 2016(6):Cd002787
Goldshtein M, Lerner O, Kalaora R, Yarkoni S, Dekel E, Confino H et al (2020) Nitric oxide is a powerful anti-coronavirus-inhaled agent that acts within hours. Chest 158(4):A2446–A2447
Good SS, Moussa A, Zhou X-J, Pietropaolo K, Sommadossi J-P (2020) 1265. AT-527, an oral purine nucleotide prodrug exhibiting potent in vitro antiviral activity against human coronaviruses, including SARS-CoV-2. Open Forum Infect Dis 7(Suppl 1):S649
Good SS, Moussa A, Zhou XJ, Pietropaolo K, Sommadossi JP (2020) Preclinical evaluation of AT-527, a novel guanosine nucleotide prodrug with potent, pan-genotypic activity against hepatitis C virus. PLoS One 15(1):e0227104
Good SS, Westover J, Jung KH, Zhou XJ, Moussa A, La Colla P et al (2021) AT-527, a double prodrug of a guanosine nucleotide analog, is a potent inhibitor of SARS-CoV-2 in vitro and a promising oral antiviral for treatment of COVID-19. Antimicrob Agents Chemother 65(4)
Gowen BB, Wong MH, Jung KH, Sanders AB, Mitchell WM, Alexopoulou L et al (2007) TLR3 is essential for the induction of protective immunity against Punta Toro Virus infection by the double-stranded RNA (dsRNA), poly(I:C12U), but not Poly(I:C): differential recognition of synthetic dsRNA molecules. J Immunol 178(8):5200–5208
Grollman AP (1966) Structural basis for inhibition of protein synthesis by emetine and cycloheximide based on an analogy between ipecac alkaloids and glutarimide antibiotics. Proc Natl Acad Sci USA 56(6):1867–1874
Harbig A, Mernberger M, Bittel L, Pleschka S, Schughart K, Steinmetzer T et al (2020) Transcriptome profiling and protease inhibition experiments identify proteases that activate H3N2 influenza A and influenza B viruses in murine airways. J Biol Chem 295(33):11388–11407
Hedenstierna G, Chen L, Hedenstierna M, Lieberman R, Fine DH (2020) Nitric oxide dosed in short bursts at high concentrations may protect against Covid 19. Nitric Oxide 103:1–3
Ho CL, Wang JL, Lee CC, Cheng HY, Wen WC, Cheng HH et al (2014) Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed Pharmacother 68(8):1007–1014
Hopp M-T, Domingo-Fernández D, Gadiya Y, Detzel MS, Schmalohr BF, Steinbock F et al (2020) Unravelling the debate on heme effects in COVID-19 infections. bioRxiv 2020.06.09.142125
Huang D, Yu H, Wang T, Yang H, Yao R, Liang Z (2021) Efficacy and safety of umifenovir for coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis. J Med Virol 93(1):481–490
Huang H, Guan L, Yang Y, Grange JML, Tang G, Xu Y et al (2020) Chloroquine, arbidol (umifenovir) or lopinavir/ritonavir as the antiviral monotherapy for COVID-19 patients: a retrospective cohort study. Res Square
Hwang L, Ng K, Wang W, Trieu V (2016) OT-101: An anti-TGF-beta-2 antisense- primed tumors to subsequent chemotherapies. J Clin Oncol 34:e1527
Ichinohe T, Ainai A, Tashiro M, Sata T, Hasegawa H (2009) PolyI:polyC12U adjuvant-combined intranasal vaccine protects mice against highly pathogenic H5N1 influenza virus variants. Vaccine 27(45):6276–6279
Idris MM, Banu S, Siva AB, Nagaraj R (2020) Downregulation of defensin genes in SARS-CoV-2 infection. medRxiv 2020.09.21.20195537
Jafarzadeh A, Nemati M (2021) Jafarzadeh S (2021) Contribution of STAT3 to the pathogenesis of COVID-19. Microb Pathog 154:104836
Jomah S, Asdaq SMB, Al-Yamani MJ (2020) Clinical efficacy of antivirals against novel coronavirus (COVID-19): a review. J Infect Public Health 13(9):1187–1195
Kang YL, Chou YY, Rothlauf PW, Liu Z, Soh TK, Cureton D et al (2020) Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci USA 117(34):20803–20813
Kelleni MT (2020) Nitazoxanide/azithromycin combination for COVID-19: a suggested new protocol for early management. Pharmacol Res 157:104874
Keyaerts E, Vijgen L, Chen L, Maes P, Hedenstierna G, Van Ranst M (2004) Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. Int J Infect Dis 8(4):223–226
Kumar R, Afsar M, Khandelwal N, Chander Y, Riyesh T, Dedar RK et al (2021) Emetine suppresses SARS-CoV-2 replication by inhibiting interaction of viral mRNA with eIF4E. Antiviral Res 189:105056
Li PA, Howlett W, He QP, Miyashita H, Siddiqui M, Shuaib A (1998) Postischemic treatment with calpain inhibitor MDL 28170 ameliorates brain damage in a gerbil model of global ischemia. Neurosci Lett 247(1):17–20
Li X, Geng M, Peng Y, Meng L, Lu S (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10(2):102–108
Li Y, Xie Z, Lin W, Cai W, Wen C, Guan Y et al (2020) Efficacy and Safety of lopinavir/ritonavir or arbidol in adult patients with mild/moderate COVID-19: an exploratory randomized controlled trial. Medicine (N Y) 1(1):105–13.e4
Lian N, Xie H, Lin S, Huang J, Zhao J, Lin Q (2020) Umifenovir treatment is not associated with improved outcomes in patients with coronavirus disease 2019: a retrospective study. Clin Microbiol Infect 26(7):917–921
Liu Q, Fang X, Tian L, Chung U, Chen X, Wang K et al (2020) The effect of arbidol hydrochloride on reducing mortality of Covid-19 patients: a retrospective study of real-world data from three hospitals in Wuhan. medRxiv 2020.04.11.20056523.
Liu Q, Xiong HR, Lu L, Liu YY, Luo F, Hou W et al (2013) Antiviral and anti-inflammatory activity of arbidol hydrochloride in influenza A (H1N1) virus infection. Acta Pharmacol Sin 34(8):1075–1083
Lokhande AS, Devarajan PV (2021) A review on possible mechanistic insights of nitazoxanide for repurposing in COVID-19. Eur J Pharmacol 891:173748
Lolli ML, Sainas S, Pippione AC, Giorgis M, Boschi D, Dosio F (2018) Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent Pat Anticancer Drug Discov 13(1):86–105
Lotz C, Muellenbach RM, Meybohm P, Mutlak H, Lepper PM, Rolfes CB et al (2021) Effects of inhaled nitric oxide in COVID-19-induced ARDS—is it worthwhile? Acta Anaesthesiol Scand 65(5):629–632
Mahshid A, Hesamoddin H (2020) Molecular mechanisms of galidesivir as a potential antiviral treatment for COVID-19. J Pharm Care 8(3)
Makowka L, Sher LS, Cramer DV (1993) The development of Brequinar as an immunosuppressive drug for transplantation. Immunol Rev 136:51–70
Mall R, Elbasir A, Almeer H, Islam Z, Kolatkar PR, Chawla S et al (2021) A modelling framework for embedding-based predictions for compound-viral protein activity. Bioinformatics 37(17):2544–2555
Martinez MA (2021) Plitidepsin: a repurposed drug for the treatment of COVID-19. Antimicrob Agents Chemother 65(4)
Martins-Filho PR, Barreto-Alves JA, Fakhouri R (2020) Potential role for nitazoxanide in treating SARS-CoV-2 infection. Am J Physiol Lung Cell Mol Physiol 319(1):L35–L36
Maury PW (2021) Targeting the receptor AXL by bemcentinib prevents SARS-CoV-2 infection. Accessed March 6, 2021
Mir JM, Maurya RC (2020) Nitric oxide boosters as defensive agents against COVID-19 infection: an opinion. J Biomol Struct Dyn 1–7
Mitchell WM (2016) Efficacy of rintatolimod in the treatment of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). Expert Rev Clin Pharmacol 9(6):755–770
Neris RLS, Figueiredo CM, Higa LM, Araujo DF, Carvalho CAM, Verçoza BRF et al (2018) Co-protoporphyrin IX and Sn-protoporphyrin IX inactivate Zika, Chikungunya and other arboviruses by targeting the viral envelope. Sci Rep 8(1):9805
Nojomi M, Yassin Z, Keyvani H, Makiani MJ, Roham M, Laali A et al (2020) Effect of arbidol (umifenovir) on COVID-19: a randomized controlled trial. BMC Infect Dis 20(1):954
Painter WP, Holman W, Bush JA, Almazedi F, Malik H, Eraut N et al (2021) Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob Agents Chemother 65(5)
Pally C, Smith D, Jaffee B, Magolda R, Zehender H, Dorobek B et al (1998) Side effects of brequinar and brequinar analogues, in combination with cyclosporine, in the rat. Toxicology 127(1–3):207–222
Pécheur EI, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M et al (2016) The synthetic antiviral drug arbidol inhibits globally prevalent pathogenic viruses. J Virol 90(6):3086–3092
Peters GJ, Sharma SL, Laurensse E, Pinedo HM (1987) Inhibition of pyrimidine de novo synthesis by DUP-785 (NSC 368390). Invest New Drugs 5(3):235–244
Ramasamy S, Subbian S (2021) Critical determinants of cytokine storm and type I interferon response in COVID-19 pathogenesis. Clin Microbiol Rev 34(3)
Reiss CS, Komatsu T (1998) Does nitric oxide play a critical role in viral infections? J Virol 72(6):4547–4551
Rele S (2020) Emerging outbreaks and epidemic threats: the practicality and limitations in the development and manufacturing of treatments for coronavirus (covid-19). Polymorphism 4:45–52
Ricciardolo FLM, Bertolini F, Carriero V, Högman M (2020) Nitric oxide’s physiologic effects and potential as a therapeutic agent against COVID-19. J Breath Res 15(1):014001
Riva L, Yuan S, Yin X, Martin-Sancho L, Matsunaga N, Burgstaller-Muehlbacher S et al (2020) A large-scale drug repositioning survey for SARS-CoV-2 antivirals. bioRxiv: the preprint server for biology 2020.04.16.044016
Riva L, Yuan S, Yin X, Martin-Sancho L, Matsunaga N, Pache L et al (2020) Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586(7827):113–119
Rocco PRM, Silva PL, Cruz FF, Junior M, Tierno P, Moura MA et al (2021) Early use of nitazoxanide in mild Covid-19 disease: randomised, placebo-controlled trial. Eur Respir J
Rosenke K, Hansen F, Schwarz B, Feldmann F, Haddock E, Rosenke R et al (2021) Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat Commun 12(1):2295
Rudrapal M, Khairnar SJ, Jadhav AG (2020) Drug repurposing (DR): an emerging approach in drug discovery. drug repurposing-hypothesis, molecular aspects and therapeutic applications IntechOpen
Salata C, Calistri A, Parolin C, Baritussio A, Palù G (2017) Antiviral activity of cationic amphiphilic drugs. Expert Rev Anti Infect Ther 15(5):483–492
Senapati S, Banerjee P, Bhagavatula S, Kushwaha PP, Kumar S (2021) Contributions of human ACE2 and TMPRSS2 in determining host-pathogen interaction of COVID-19. J Genet 100(1)
Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML et al (2020) An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med 12(541)
Shen L, Niu J, Wang C, Huang B, Wang W, Zhu N et al (2019) High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J Virol 93(12):e00023-e119
Strayer DR, Carter WA, Stouch BC, Stevens SR, Bateman L, Cimoch PJ et al (2012) A double-blind, placebo-controlled, randomized, clinical trial of the TLR-3 agonist rintatolimod in severe cases of chronic fatigue syndrome. PLoS One 7(3):e31334
Tian L, Qiang T, Liang C, Ren X, Jia M, Zhang J et al (2021) RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur J Med Chem 213:113201
Trougakos IP, Stamatelopoulos K, Terpos E, Tsitsilonis OE, Aivalioti E, Paraskevis D et al (2021) Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J Biomed Sci 28(1):9
Uckun F, Hwang L, Trieu V (2020) Selectively targeting TGF-2 with Trabedersen/OT-101 in treatment of evolving and mild ards inCOVID-19. Clin Investig 10:35–44
Uckun FM, Trieu V (2020) Targeting transforming growth factor-beta for treatment of COVID-19-associated Kawasaki disease in children. Clin Res Pediatr 3(1):1–3
Uludağ H, Parent K, Aliabadi HM, Haddadi A (2020) Prospects for RNAi therapy of COVID-19. Front Bioeng Biotechnol 8:916
Wang A, Sun Y, Liu Q, Wu H, Liu J, He J et al (2020) Low dose of emetine as potential anti-SARS-CoV-2 virus therapy: preclinical in vitro inhibition and in vivo pharmacokinetic evidences. Molecular Biomedicine 1(1):14
Wang X, Cao R, Zhang H, Liu J, Xu M, Hu H et al (2020) The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov 6:28
Warren TK, Wells J, Panchal RG, Stuthman KS, Garza NL, Van Tongeren SA et al (2014) Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508(7496):402–405
Wewers MD, Crystal RG (2013) Alpha-1 antitrypsin augmentation therapy. COPD 10(Suppl 1):64–67
White KM, Rosales R, Yildiz S, Kehrer T, Miorin L, Moreno E et al (2021) Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 371(6532):926–931
Wiegand SB, Safaee Fakhr B, Carroll RW, Zapol WM, Kacmarek RM, Berra L (2020) Rescue treatment with high-dose gaseous nitric oxide in spontaneously breathing patients with severe coronavirus disease 2019. Crit Care Explor 2(11):e0277
Wilkinson T, Dixon R, Page C, Carroll M, Griffiths G, Ho LP et al (2020) ACCORD: a multicentre, seamless, phase 2 adaptive randomisation platform study to assess the efficacy and safety of multiple candidate agents for the treatment of covid-19 in hospitalised patients: a structured summary of a study protocol for a randomised controlled trial. Trials 21(1):691
Xiong R, Zhang L, Li S, Sun Y, Ding M, Wang Y et al (2020) Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 11(10):723–739
Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A et al (2020) Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 9(1):761–770
Xu K, Chen Y, Yuan J, Yi P, Ding C, Wu W et al (2020) Clinical efficacy of arbidol in patients with 2019 novel coronavirus-infected pneumonia: a retrospective cohort study. SSRN J
Xu P, Huang J, Fan Z, Huang W, Qi M, Lin X et al (2020) Arbidol/IFN-α2b therapy for patients with corona virus disease 2019: a retrospective multicenter cohort study. Microbes Infect 22(4–5):200–205
Xu Y, Jiang H (2020) Potential treatment of COVID-19 by inhibitors of human dihydroorotate dehydrogenase. Protein Cell 11(10):699–702
Yang C, Chapman KR, Wong A, Liu M (2021) α1-Antitrypsin deficiency and the risk of COVID-19: an urgent call to action. Lancet Respir Med 9(4):337–339
Yang C, Ke C, Yue D, Li W, Hu Z, Liu W et al (2020) Effectiveness of arbidol for COVID-19 prevention in health professionals. Front Public Health 8:249
Yang C, Keshavjee S, Liu M (2020) Alpha-1 antitrypsin for COVID-19 treatment: dual role in antiviral infection and anti-inflammation. Front Pharmacol 11:615398
Yang S, Xu M, Lee EM, Gorshkov K, Shiryaev SA, He S et al (2018) Emetine inhibits Zika and Ebola virus infections through two molecular mechanisms: inhibiting viral replication and decreasing viral entry. Cell Discov 4:31
Yu CC, Chiang PC, Lu PH, Kuo MT, Wen WC, Chen P et al (2012) Antroquinonol, a natural ubiquinone derivative, induces a cross talk between apoptosis, autophagy and senescence in human pancreatic carcinoma cells. J Nutr Biochem 23(8):900–907
Zamanian RT, Pollack CV Jr, Gentile MA, Rashid M, Fox JC, Mahaffey KW et al (2020) Outpatient inhaled nitric oxide in a patient with vasoreactive idiopathic pulmonary arterial hypertension and COVID-19 infection. Am J Respir Crit Care Med 202(1):130–132
Zarandi PK, Zinatizadeh MR, Zinatizadeh M, Yousefi MH, Rezaei N (2021) SARS-CoV-2: from the pathogenesis to potential anti-viral treatments. Biomed Pharmacother 137:111352
Zheng L, Zhang L, Huang J, Nandakumar KS, Liu S, Cheng K (2020) Potential treatment methods targeting 2019-nCoV infection. Eur J Med Chem 205:112687
Zhu Z, Lu Z, Xu T, Chen C, Yang G, Zha T et al (2020) Arbidol monotherapy is superior to lopinavir/ritonavir in treating COVID-19. J Infect 81(1):e21–e23
Funding
This research did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest related to this work.
Additional information
Handling Editor: Pablo Pineyro.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Beheshtirouy, S., Khani, E., Khiali, S. et al. Investigational antiviral drugs for the treatment of COVID-19 patients. Arch Virol 167, 751–805 (2022). https://doi.org/10.1007/s00705-022-05368-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05368-z