
Abstract
Hirame rhabdovirus (HIRRV), a member of the genus Novirhabdovirus, causes morbidity and mortality in farmed olive flounder (Paralichthys olivaceus). As no information is available on the role of the NV gene of HIRRV, we produced a recombinant HIRRV with the NV gene deleted (rHIRRV-ΔNV) using reverse genetic technology and investigated whether the NV gene knockout affected HIRRV replication and the type I interferon response of the host cell. The rescue of rHIRRV-ΔNV was successful only when IRF9-gene-knockout Epithelioma papulosum cyprini (ΔIRF9-EPC) cells were used, suggesting that the NV protein of HIRRV might be involved in inhibition of the type I interferon response of the host cell. This conclusion was also supported by the significantly higher level of Mx gene induction in EPC cells infected with rHIRRV-ΔNV than in cells infected with recombinant HIRRV without the deletion. When cells were coinfected with rHIRRV-ΔNV and either wild-type HIRRV or wild-type viral hemorrhagic septicemia virus (VHSV), there was a decrease in the growth rate of not only wild-type HIRRV but also wild-type VHSV in a concentration-dependent manner. Further studies are required to investigate the role of HIRRV NV in virulence and its possible importance for the development of attenuated vaccines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hirame rhabdovirus (HIRRV) is a negative-sense, single-stranded RNA virus that belongs to the genus Novirhabdovirus, family Rhabdoviridae. The genome of HIRRV is approximately 11 kb in length and contains six open reading frames (ORFs) encoding a nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), non-virion (NV) protein, and RNA-dependent RNA polymerase (L) in the order N-P-M-G-NV-L [1]. HIRRV was first isolated form olive flounder (Paralichthys olivaceus) and ayu (Plecoglossus altivelis) in Japan [2] and is also an important pathogen in cultured olive flounder in Korea and China [3,4,5]. However, recently, HIRRV was discovered in freshwater fish species in Europe [6] and in other cultured marine fish species including the blackhead seabream (Acanthopagrus schlegelii) and sea bass (Lateolabrax maculatus) in Korea [7, 8]. In view of the high rate of variation in RNA viruses, the possibility of HIRRV infecting various fish species and causing damage on aquaculture farms cannot be excluded, and more information about HIRRV is therefore needed.
The ability to generate recombinant viruses using reverse genetic technology has allowed researchers to investigate the functions of individual viral genes and to produce attenuated viruses [9, 10]. The presence of an NV gene is a peculiar characteristic of novirhabdoviruses [11], and the functional role of the NV protein of novirhabdoviruses has been studied using NV gene-knockout recombinant viruses. Thoulouze et al. [12] reported that a NV gene-knockout infectious hematopoietic necrosis virus (rIHNV-ΔNV) showed severely retarded growth. However, the replication of rIHNV-ΔNV was restored to normal when the NV protein was provided in trans. The NV protein of viral hemorrhagic septicemia virus (VHSV) has been reported to inhibit type I interferon responses, apoptosis, and NF-κB activation [13,14,15,16]. Moreover, due to its lowered viral replication ability and weakened pathogenicity [13, 17], NV gene-knockout VHSV has been suggested as a potential attenuated vaccine [18, 19]. However, Johnson et al. [20] and Alonso et al. [21] reported that, unlike those of VHSV and IHNV, the NV gene of snakehead rhabdovirus (SHRV) had no effect on replication and virulence, although they pointed out a possible spacing role of the SHRV NV gene in regulation of transcription through the G/NV junction.
To investigate the function of the HIRRV NV gene, we produced a recombinant HIRRV with the NV gene deleted (rHIRRV-ΔNV) using reverse genetic technology and analyzed whether the NV gene knockout affected HIRRV replication and the type I interferon response of the host cell. The results clearly showed that the growth rate of HIRRV was severely reduced and the type I interferon response against HIRRV infection in the host cell was significantly increased by the NV gene knockout.
Materials and methods
Cells and viruses
Epithelioma papulosum cyprini (EPC) cells were grown in Leibovitz medium (L-15, Sigma) enriched with 10% fetal bovine serum (FBS, Welgene) and penicillin-streptomycin (Welgene). Interferon regulatory factor 9 (IRF9) gene knockout EPC (ΔIRF9-EPC) cells established in our previous study [22] were also cultured under the same conditions as EPC cells.
HIRRV CA 9703 [3, 23] was propagated in EPC cells at 15 ℃ in the presence of 2% FBS. When the cells showed a marked cytopathic effect (CPE), the supernatant was collected, passed through a 0.45-μm syringe filter (Advantec), and used as the viral stock. Viral hemorrhagic septicemia virus (VHSV) KJ2008 was also grown and stored in the same manner.
Generation of recombinant HIRRV
To rescue a recombinant HIRRV with the same genome sequence as wild-type HIRRV CA 9703 (GenBank accession no. AF104985.2), total RNA was extracted from EPC cells infected with wild-type HIRRV using TRIzol Reagent (Invitrogen), treated with DNase I using a Riboclear Plus Kit (GeneAll, Korea), and converted to cDNA using a Reverse Transcription System (Promega) according to the manufacturer’s instructions. The antigenomic cDNA of HIRRV was divided into eight fragments, and each fragment was amplified by PCR using the primer sets listed in Table 1. A vector backbone fragment containing a T7 promoter and a hepatitis delta virus ribozyme was amplified by PCR using the previously described construct pVHSV-wild [17] as a template to insert the HIRRV antigenomic cDNA between the promoter and the ribozyme. After the verification of the sequences, all the fragments were assembled step by step using Overlap Cloner (ELPIS, Korea) according to the manufacturer’s instructions, and the resulting construct was designated as pHIRRV-wild. To make helper constructs, the N, P, and L genes were amplified by PCR using pHIRRV-wild as a template and cloned into pGEM-T-Easy Vector and then ligated to the pFC vector (SystemBio) using the restriction enzymes AgeI and NotI for the N and P genes and AfeI and NotI for the L gene, resulting in the recombinant plasmids pCMV-N, pCMV-P, and pCMV-L. All primers used to make these constructs are listed in Table 1. EPC cells expressing T7 RNA polymerase were transfected with a mixture of pHIRRV-wild (2000 ng), pCMV-N (500 ng), pCMV-P (300 ng), and pCMV-L (200 ng) using Fugene HD transfection reagent (Promega). To test whether the N, P, and L genes of VHSV were able to rescue recombinant HIRRV, EPC cells expressing T7 RNA polymerase were co-transfected with pHIRRV-wild and VHSV helper constructs that were described previously [17]. When an extensive cytopathic effect (CPE) was observed, the supernatant was collected, passed through a 0.45-μm syringe filter, and then passaged several times on EPC cells to increase the titer of the recombinant virus.
To investigate the function of the NV gene of HIRRV, a HIRRV cDNA construct lacking the NV gene was made by combining two fragments (excluding the NV gene ORF) that had been amplified by PCR using the pHIRRV-wild construct as a template and the primers shown in Table 1, and it was designated as pHIRRV-ΔNV. EPC cells expressing T7 RNA polymerase were co-transfected with pHIRRV-ΔNV and the helper constructs for HIRRV, and the passage of rHIRRV-ΔNV was done by inoculation of ΔIRF9-EPC cells. The generation of rHIRRV-ΔNV was verified by reverse transcription PCR (RT-PCR) using the primers shown in Table 1.
Virus titration using a plaque assay
The viral titer was determined by plaque assay [24]. EPC cells and △IRF9-EPC cells (2 × 106 cells/35-mm dish) were infected with serially diluted (10-3 to 10-5) viral stock of rHIRRV-wild and rHIRRV-ΔNV, respectively, for 2 h at 15 ℃. After incubation, the medium was removed and the dishes were overlaid with plaque medium (0.7% agarose in L-15 medium containing 2% FBS, and 100 U of penicillin, and 100 μg of streptomycin per ml), and titers were determined as described by Kim et al. [17].
Comparison of rHIRRV-ΔNV growth in EPC cells and △IRF9-EPC cells
The growth of rHIRRV-ΔNV in EPC cells and △IRF9-EPC cells was analyzed using a plaque assay. Cells (3 × 106 cells/35-mm dish) were infected with rHIRRV-ΔNV at an MOI of 0.0001, and supernatant was sampled at 1, 3, 5, 7, and 9 days postinfection for plaque assay.
Effect of rHIRRV on the type I interferon response
To measure the effect of rHIRRV infection on the type I interferon response, EPC cells transfected with the plasmid pOFMx [14], which contains a luciferase reporter gene under control of an the olive flounder Mx promoter, were infected with wild-type HIRRV, rHIRRV-wild, or rHIRRV-△NV at an MOI of 0.01. At 12, 24, 36, and 48 h postinfection, the luciferase activity was measured as described by Kim and Kim [14].
Effect of rHIRRV-△NV coinfection on the replication of wild-type HIRRV and wild-type VHSV
EPC cells (3 × 106 cells/35-mm dish) were infected with wild-type HIRRV alone or wild-type VHSV alone at an MOI of 0.00001 or coinfected with each wild-type virus (MOI 0.00001) plus rHIRRV-△NV (MOI 0.01, 0.001, and 0.0001) and incubated at 15°C. To investigate the effect of rHIRRV-△NV on the replication of wild-type viruses, each supernatant was collected at 1, 3, and 5 days postinfection, and a plaque assay was performed.
Statistical analysis
Data pertaining to induction of Mx gene expression by recombinant HIRRVs were analyzed by one-way analysis of variance (ANOVA) followed by the Tukey HSD post-hoc test, using GraphPad Prism (GraphPad Software, Inc., USA). Differences between groups were considered to be significant at P < 0.05.
Results
Generation of recombinant HIRRV
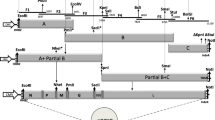
EPC cells co-transfected with pHIRRV-wild (Fig. 1) and helper constructs for HIRRV or VHSV showed clear CPE, and the rescued rHIRRV-wild induced strong CPE within 3-4 days even in the first passage on EPC cells. Whereas EPC cells co-transfected with pHIRRV-ΔNV and helper constructs did not show any clear CPE and the supernatant isolated from the transfected EPC cells also did not induce any distinct CPE, distinct CPE was observed when IRF9 gene knockout EPC (△IRF9-EPC) cells were exposed to the supernatant, and rHIRRV-ΔNV could be rescued.

Schematic representation of plasmid constructs used for the generation of a recombinant wild-type HIRRV (pHIRRV-wild) and a recombinant HIRRV lacking the NV gene (pHIRRV-ΔNV). T7p, T7 RNA polymerase promoter; HDV, hepatitis delta virus ribozyme
To verify that rHIRRV-ΔNV was successfully produced, RT-PCR was performed. Regions extending from the P gene to the G gene, from the G gene to the NV gene, and the NV gene itself were successfully amplified from the rHIRRV-wild stock, whereas the regions ranging from the G gene to the NV gene and the NV gene itself were not amplified from the stock of rHIRRV-ΔNV (Fig. 2).

Verification of the NV gene deletion in the genome of rHIRRV-ΔNV by RT-PCR. The regions from the P gene to the G gene (P-G; 1289 bp), from the G gene to the NV gene (G-NV; 1054 bp), and the NV gene (335 bp) were amplified from rHIRRV-wild stock (Left), whereas the regions from the G gene to the NV gene and the NV gene were not amplified from the rHIRRV-ΔNV stock, which lacks the NV gene (Right). M, 1kb DNA ladder; NC; negative control
Replication ability of rHIRRV-ΔNV
In EPC cells, rHIRRV-ΔNV did not show evident CPE until 9 days postinfection. However, in ΔIRF9-EPC cells, rHIRRV-ΔNV induced distinct CPE, and almost all cells were lysed at 9 days postinfection (Fig. 3A).

(A) The induction of a cytopathic effect (CPE) by the infection of naive EPC cells (EPC) and IRF9 gene knockout EPC cells (ΔIRF9) with rHIRRV-ΔNV. CPE was observed at 1, 3, 5, 7, and 9 days postinfection. (B) Growth (plaque number, pfu) of rHIRRV-ΔNV in naive EPC cells (EPC) and IRF9-gene-knockout EPC cells (ΔIRF9-EPC)
An analysis of viral growth using a plaque assay showed that the titers of rHIRRV-ΔNV cultured in EPC cells increased very slowly, whereas the titers in ΔIRF9-EPC cells increased much more rapidly (Fig. 3B).
Effect of rHIRRV-ΔNV infection on Mx gene expression in EPC cells
The type I interferon response of EPC cells in response to infection with the recombinant HIRRV was analyzed using cells harboring a plasmid construct containing an Mx-gene-promoter-driven luciferase expression cassette. The luciferase activity in cells infected with rHIRRV-ΔNV was significantly higher than that in cells infected with wild-type HIRRV or rHIRRV-wild (Fig. 4).

Effect of wild-type HIRRV, rHIRRV-wild, and rHIRRV-ΔNV infection on the luciferase activity of EPC cells containing a reporter construct with a Mx-gene-promoter-driven luciferase expression cassette. The asterisks on the bar represent statistical significance at p < 0.05.
Effect of rHIRRV-ΔNV coinfection on the replication of wild-type HIRRV and wild-type VHSV
To analyze the effect of rHIRRV-ΔNV coinfection on wild-type HIRRV and wild-type VHSV replication, EPC cells were coinfected with each wild-type virus plus various concentrations of rHIRRV-ΔNV. Then, at 1, 3, and 5 days postinfection, a plaque assay was performed to measure the viral titer. Cells coinfected with each wild-type virus and rHIRRV-ΔNV showed a slow progression of CPE compared to cells infected with each wild-type virus alone, and this effect was dependent on the concentration of rHIRRV-ΔNV (Fig. 5). The plaque assay data also demonstrated inhibition of the growth of wild-type HIRRV (Fig. 6A) and wild-type VHSV (Fig. 6B) by coinfection with rHIRRV-ΔNV in a concentration-dependent manner.

Effect of rHIRRV-ΔNV (MOI 0.01, 0.001, and 0.0001) on the replication of wild-type HIRRV (A) and wild-type VHSV (B) in EPC cells. The progression of CPE was observed at 1, 3, 5, 7, and 9 days postinfection.

Effect of rHIRRV-ΔNV (ΔNV; MOI 0.01, 0.001, and 0.0001) on the growth of wild-type HIRRV (wtH) (A) and wild-type VHSV (wtV) (B) in EPC cells. The virus titer was determined by plaque assay.
Discussion
In the present study, for the first time, we rescued an NV-gene-deleted recombinant HIRRV using reverse genetic technology. Although rHIRRV-wild was successfully generated from EPC cells transfected with full genome and helper constructs, rHIRRV-ΔNV was not efficiently rescued from EPC cells transfected with the corresponding constructs, suggesting that the deletion of the NV gene might play an important role in the replication of HIRRV. The role of the NV gene of novirhabdoviruses has been investigated mainly using VHSV and IHNV, and the inhibition of the host cell’s type I interferon response by NV protein was verified in those studies [14, 25, 26]. In this study, we hypothesized that the NV protein of HIRRV would also act as an inhibitor of the type I interferon response. In our previous study, we established an IRF9 gene knockout EPC (ΔIRF9-EPC) cell line using a CRISPR/Cas9 system to disable the type I interferon response of the host [22]. Therefore, in this study, we used ΔIRF9-EPC cells to rescue rHIRRV-ΔNV. In view of the critical role of the IRF9 gene in the induction of the type I interferon response, the present result indicates that the NV protein of HIRRV is involved in the inhibition of the host cell’s type I interferon response. This is also supported by the observation of significantly higher Mx1 gene induction in EPC cells infected with rHIRRV-ΔNV than in cells infected with rHIRRV-wild.
In experiments investigating the effect of rHIRRV-ΔNV on the growth of wild-type HIRRV, wild-type HIRRV showed decreased growth when co-cultured with rHIRRV-ΔNV, and the inhibitory activity of rHIRRV-ΔNV was dependent on the titer of rHIRRV-ΔNV. Defective interfering particles (DIPs) of RNA viruses differ from standard infectious viral particles in that they have a defective viral genome, which makes them able to replicate only when they are helped by coinfection with intact virus [27]. Thus, recombinant viruses with non-functional essential genes for replication can be included in DIPs [28]. Kim et al. [29] reported that G-gene-deleted single-cycle VHSV (rVHSV-ΔG) inhibited the growth of wild-type VHSV by co-infection and that rVHSV-ΔG could be regarded as a DIP. However, the present rHIRRV-ΔNV differs from DIPs because it can replicate without the help of intact viruses. Therefore, in the present study, the inhibition of wild-type HIRRV growth by coinfection with rHIRRV-ΔNV might have been caused by the sharing of N, P, L proteins between wild-type HIRRV and rHIRRV-ΔNV. As N, P, and L proteins are essential for genome replication and viral gene transcription, the use of a portion of those proteins for the slowly replicating rHIRRV-ΔNV might decrease the pool of proteins available for wild-type HIRRV replication, leading to the retarded growth of wild-type HIRRV. In the present study, rHIRRV-ΔNV decreased the growth not only of wild-type HIRRV but also of wild-type VHSV. This heterologous inhibition by rHIRRV-ΔNV can be explained by our preliminary experimental results, in which rHIRRV-wild could be rescued from cells transfected with the HIRRV full genome construct and VHSV N, P, L helper constructs. Therefore, as with wild-type HIRRV, the VHSV N, P, and L proteins might be diverted for the replication of rHIRRV-ΔNV, resulting in slower growth of wild-type VHSV. Furthermore, as the infection was carried out at a low MOI, it cannot be excluded that type I interferon responses elicited by the first round of infection with rHIRRV-ΔNV might have inhibited the multiplication of wild-type HIRRV and VHSV.
In conclusion, a NV-gene-deleted recombinant HIRRV was rescued using reverse genetics, and the inhibitory role of the HIRRV NV gene on the type I interferon response of the host cell was demonstrated. Further studies investigating the applicability of recombinant HIRRV for the analysis of the mechanism of virulence and for the development of attenuated vaccines are required.
References
Walker PJ, Dietzgen RG, Joubert DA (2011) Rhabdovirus accessory genes. Virus Res 162:110–125
Kimura T, Yoshimizu M, Gorie S (1986) A new rhabdovirus isolated in Japan from cultured hirame (Japanese flounder) Paralichthys olivaceus and ayu Plecoglossus altivelis. Dis Aquat Org 1:209–217
Oh MJ, Choi TJ (1998) A new rhabdovirus (HRV-like) isolated in Korea from cultured Japanese flounder Paralichthys olivaceus. J Fish Pathol 11:129–136
Yingjie S, Min Z, Hong L (2011) Analysis and characterization of the complete genomic sequence of the Chinese strain of hirame rhabdovirus. J Fish Dis 34:167–171
Zhang J, Tang X, Sheng X et al (2017) Isolation and identification of a new strain of hirame rhabdovirus (HIRRV) from Japanese flounder Paralichthys olivaceus in China. Virol J 14:73
Borzym E, Matras M, Majpaluch J, Baud M (2014) First isolation of hirame rhabdovirus from freshwater fish in Europe. J Fish Dis 37:423–430
Kim WS, Oh MJ (2015) Hirame rhabdovirus (HIRRV) as the cause of a natural disease outbreak in cultured black seabream (Acanthopagrus schlegeli) in Korea. Arch Virol 160:3063–3066
Seo HG, Do JW, Jung SH (2016) Han HJ (2016) Outbreak of hirame rhabdovirus infection in cultured spotted sea bass Lateolabrax maculatus on the western coast of Korea. J Fish Dis 39:1239–1246
Biacchesi S (2011) The reverse genetics applied to fish RNA viruses. Vet Res 42:12
Pfaller CK, Cattaneo R, Schnell MJ (2015) Reverse genetics of Mononegavirales: how they work, new vaccines, and new cancer therapeutics. Virology 479–480:331–344
Kurath G, Higman KH, Bjorklund HV (1997) Distribution and variation of NV genes in fish rhabdoviruses. J Gen Virol 78:113–117
Thoulouze MI, Bouguyon E, Carpentier C, Brémont M (2004) Essential role of the NV protein of Novirhabdovirus for pathogenicity in rainbow trout. J Virol 78:4098–4107
Ammayappan A, Vakharia VN (2011) Nonvirion protein of Novirhabdovirus suppresses apoptosis at the early stage of virus infection. J Virol 85:8393–8402
Kim MS, Kim KH (2012) Effects of NV gene knock-out recombinant viral hemorrhagic septicemia virus (VHSV) on Mx gene expression in Epithelioma papulosum cyprini (EPC) cells and olive flounder (Paralichthys olivaceus). Fish Shellfish Immunol 32:459–463
Kim MS, Kim KH (2013) The role of viral hemorrhagic septicemia virus (VHSV) NV gene in TNF-α- and VHSV infection-mediated NF-κB activation. Fish Shellfish Immunol 34:1315–1319
Chinchilla B, Gomez-Casado E (2017) Identification of the functional regions of the viral haemorrhagic septicaemia virus (VHSV) NV protein: Variants that improve function. Fish Shellfish Immunol 70:343–350
Kim MS, Kim DS, Kim KH (2011) Generation and characterization of NV gene-knockout recombinant viral hemorrhagic septicemia virus (VHSV) genotype IVa. Dis Aquat Org 97:25–35
Kim MS, Kim KH (2011) Protection of olive flounder, Paralichthys olivaceus, against viral hemorrhagic septicemia virus (VHSV) by immunization with NV gene-knockout recombinant VHSV. Aquaculture 314:39–43
Kim MS, Kim DS, Kim KH (2011) Oral immunization of olive flounder (Paralichthys olivaceus) with recombinant live viral hemorrhagic septicemia virus (VHSV) induces protection against VHSV infection. Fish Shellfish Immunol 31:212–216
Johnson MC, Simon BE, Kim CH, Leong JA (2000) Production of recombinant snakehead rhabdovirus: the NV protein is not required for viral replication. J Virol 74:2343–2350
Alonso M, Kim CH, Johnson MC, Pressley M, Leong JA (2004) The NV gene of snakehead rhabdovirus (SHRV) is not required for pathogenesis, and a heterologous glycoprotein can be incorporated into the SHRV envelope. J Virol 78:5875–5882
Kim MS, Shin MJ, Kim KH (2018) Increase of viral hemorrhagic septicemia virus growth by knockout of IRF9 gene in Epithelioma papulosum cyprini cells. Fish Shellfish Immunol 83:443–448
Kim DH, Oh HK, Eou JI, Seo HJ, Kim SK, Oh MJ, Nam SW, Choi TJ (2005) Complete nucleotide sequence of the hirame rhabdovirus, a pathogen of marine fish. Virus Res 107:1–9
Burke JA, Mulcahy D (1980) Plaquing procedure for infectious hematopoietic necrosis virus. Appl Environ Microbiol 39:872–876
Biacchesi S, Mérour E, Chevret D, Lamoureux A, Bernard J, Brémont M (2017) NV proteins of fish Novirhabdovirus recruit cellular PPM1Bb protein phosphatase and antagonize RIG-I-mediated IFN induction. Sci Rep 7:44025
Wu Y, Wang L, Guo T, Jiang Y, Qiao X, Sun L, Liu M, Tang L, Xu Y, Li Y (2018) Identification of amino acid residues in infectious hematopoietic necrosis virus (IHNV) NV protein necessary for viral replication and pathogenicity. Fish Shellfish Immunol 79:294–302
Ziegler CM, Botten JW (2020) Defective interfering particles of negative-strand RNA viruses. Trends Microbiol 28:554–565
Roux L, Simon AE, Holland JJ (1991) Effects of defective interfering viruses on virus replication and pathogenesis in vitro and in vivo. Adv Virus Res 40:181–211
Kim MS, Choi SH, Kim KH (2016) Effect of G gene-deleted recombinant viral hemorrhagic septicemia virus (rVHSV-ΔG) on the replication of wild type VHSV in a fish cell line and in olive flounder (Paralichthys olivaceus). Fish Shellfish Immunol 54:598–601
Acknowledgements
This research was a part of the project (2020) titled ‘Development of rapid and sensitive diagnostic methods for the quarantine of aquatic animals and their products’, funded by the Ministry of Oceans and Fisheries, Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Research involving human participants and/or animals
No part of this study included experiments with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Kalpana Agnihotri.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ryu, S.J., Kwak, J.S. & Kim, K.H. Effect of NV gene deletion in the genome of hirame rhabdovirus (HIRRV) on viral replication and the type I interferon response of the host cell. Arch Virol 167, 77–84 (2022). https://doi.org/10.1007/s00705-021-05286-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05286-6