
Abstract
Porcine circovirus type 2 (PCV2) is the most ubiquitous viral pathogen of pigs and has persistently affected the global swine industry. Since first being identified in South Korea in 1999, the virus has undergone considerable genetic change and genotype shifts during the past two decades. These events have contributed to the coexistence of genotypes PCV2a, PCV2b, and PCV2d in Korean pig populations, which may promote viral recombination. The genotypic and phylogenetic characteristics of PCV2 strains circulating in pig herds on Jeju Island from 2019 to 2020 were the focus of this study. Genotype-specific PCR indicated that PCV2d is the dominant viral genotype and that coinfections with PCV2d and PCV2a (75%) or PCV2a and PCV2b (25%) are common in provincial pig herds. The complete genome sequences of 11 PCV2 strains, including three PCV2a, two PCV2b, and six PCV2d strains, were determined. A genomic comparison showed that all of the viruses had the highest nucleotide sequence identity to their corresponding genotypic reference strain. Notably, genetic and phylogenetic analysis revealed that one PCV2d strain, KNU-1931, exhibited nucleotide sequence variation in the ORF1 gene when compared to other PCV2d strains but showed a high degree of similarity to the PCV2b strains. Comprehensive recombination analysis suggested that KNU-1931 originated from natural recombination within ORF1 between PCV2b (the minor parent) and PCV2d (the major parent) strains. Our findings provide information about the frequency of genetic recombination between two different PCV2 genotypes circulating in the field domestically, illustrating the importance of continual intergenotypic recombination for viral fitness when multiple genotypes are present.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine circovirus type 2 (PCV2) was first identified in the late 1990s and was initially associated with PCV2 systemic disease (PCV2-SD) [1]. A number of pathological conditions related to PCV2 infection have been described in the last two decades, including PCV2 reproductive disease, PCV2 lung disease, PCV2 enteric disease, and porcine dermatitis and nephropathy syndrome. All PCV2-related manifestations are collectively referred to as "porcine circovirus-associated diseases" (PCVADs) [2, 3].
PCV2 is a small, nonenveloped virus with a circular, covalently closed single-stranded DNA genome and is a member of the genus Circovirus in the family Circoviridae [4]. The PCV2 genome is 1.76–1.77 kb in length and contains four open reading frames (ORFs; ORF1–ORF4). ORF1 is located on the positive strand and encodes the nonstructural replicase proteins Rep and Rep′, which are responsible for viral replication, while ORF2 is located on the complementary strand and codes for the sole structural protein, Cap, which is associated with immunogenicity [5,6,7]. ORF3 and ORF4 are embedded within ORF1 in the antisense orientation and encode nonstructural proteins that regulate virus-induced apoptosis [8, 9].
PCV2 is divided into five genotypes (PCV2a, PCV2b, PCV2c, PCV2d, and PCV2e) based on the ORF2 sequence [3, 10,11,12]. PCV2a, PCV2b, and PCV2d are the prevalent genotypes in the global pig population [7, 12, 13], while PCV2c has only been isolated from archived serum samples in Denmark [14] and feral pigs in Brazil [15]. A retrospective investigation revealed a new phylogenetic cluster that has been proposed as genotype "PCV2e" [10].
In South Korea, PCV2 was first identified in 1999 in pigs with PCV2-SD [16]. PCV2a was the initial predominant genotype until the early 2000s, but a genotype shift from PCV2a to PCV2b occurred around 2002 [17]. Furthermore, the continuous countrywide circulation of classical PCV2a and PCV2b strains resulted in the appearance of recombinant PCV2 strains via intergenotypic recombination within ORF1 [18]. A recent molecular epidemiology study indicated that a second genotype shift to PCV2d occurred nationwide before 2012 and that the coexistence of multiple genotypes (PCV2a, PCV2b, and PCV2d) is common in Korean swine herds [19]. Interestingly, the authors of that study also reported the occurrence of a genotype shift to PCV2d in Jeju Province, the largest island of South Korea, where the trade of live pigs from the mainland is not permitted [19]. In addition, infection pattern analysis at the farm level confirmed that single infections with PCV2d (57.1%) or dual infections with PCV2d (28.6%) and PCV2a or PCV2b have been frequent in this province [19]. However, despite the genotype shift, there is limited information regarding the incidence of new intergenotypic recombination of PCV2 in South Korea. Therefore, we aimed to expand our knowledge of the genetic diversity of PCV2 isolates in pig herds in Jeju Province from 2019 to 2020 based on complete genome sequences. This study provides direct evidence of an emerging novel recombinant strain that originated from a natural intergenotypic recombination event, and the genome of this strain is composed of a PCV2d backbone with a partial ORF1 sequence from of PCV2b circulating in South Korea.
Materials and methods
Collection of clinical samples
Clinical samples (blood, oral fluid, or feces) were obtained from pigs of different ages from eight commercial farrow-to-finish farms located in the Hallim district of Jeju Province from October 2019 to March 2020. The farms had been clinically affected by PCVAD-like symptoms, including respiratory disorders and wasting and were distributed within a 5-km radius (Supplementary Fig. S1). Details about the sample collection are provided in Supplementary Table S1. The collected fecal samples were diluted 1:10 (w/v) with phosphate-buffered saline. The fecal suspensions, as well as the blood and oral fluids, were centrifuged for 10 min at 4,500 × g in a Hanil Centrifuge Fleta 5 (Incheon, South Korea). The clarified supernatants and serum samples were initially subjected to real-time quantitative PCR (qPCR) analysis for the detection of PCV2 as described previously [20]. The PCV2-positive DNA samples were subjected to additional PCR to determine PCV2 genotypes using three genotype-specific primer sets (Table 1).
Nucleotide sequence analysis
The full-length genomes of 11 PCV2 strains were amplified by PCR using specific primer sets (Table 1). The PCR amplicons were gel-purified, cloned using the pGEM-T Easy Vector System (Promega, Madison, WI), and sequenced in both directions using commercial vector-specific T7 and SP6 primers. The complete genomic sequences of the PCV2 strains were deposited in the GenBank database under the accession numbers listed in Table 2. We selected ten colonies for sequencing from each pGEM-T cloning experiment to rule out the possibility of a mixed infection in a single sample, as well as to exclude any contamination that may have occurred during sample preparation.
Multiple alignments and phylogenetic analysis
The sequences of ORF2 genes and the complete genomes of 74 global PCV2 strains from the GenBank database were used to produce sequence alignments and to perform phylogenetic analysis. ClustalX 2.0 [21] was used to generate multiple sequence alignments and determine percent nucleotide sequence divergence. Phylogenetic trees were constructed from the aligned nucleotide or amino acid sequences using the neighbor-joining method and were subjected to bootstrap analysis with 1000 replicates to determine the percent reliability value for each internal node of the tree [22]. All phylogenetic trees were generated using MEGA X software [23].
Recombination analysis
Recombination events were detected using three methods. First, whole-genome sequences were aligned and analyzed using the Recombination Detection Program (RDP 4 version 4.95) to simultaneously detect potential recombination events using eight algorithms (RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq, and LARD) [24]. A PCV2 sequence was considered recombinant when the recombination signal was supported by at least four of these methods with p-values of less than 0.01 to ensure reliability. Recombination breakpoint detection by at least four methods was considered confirmation of a putative recombination event. Second, the potential recombination events and breakpoints were verified by similarity plot analysis using SimPlot version 3.5.1 [25]. Finally, the putative recombination data were supported by phylogenetic analysis of separate regions of the parental genome as described above.
Results
Real-time qPCR for PCV2 detection was performed on all samples obtained from the eight farms. PCV2-positive samples with low Ct values representing each farm were selected and used to determine the genotypes of the PCV2 strains circulating on Jeju Island using genotype-specific conventional PCR (Supplementary Table S1). As expected, PCV2d was detected in all samples from PCV2-positive farms, confirming its high prevalence in the provincial herds. Interestingly, no singly occurring PCV2d infection was discovered at the farm level. Two or more PCV2 genotypes commonly co-circulated on all eight farms; dual infections with PCV2a and PCV2d were identified on six farms, and triple infections with PCV2a, PCV2b, and PCV2d were found on two farms (Table 2).
Subsequently, we were able to determine the full-length genomic sequences of 11 PCV2 isolates: three PCV2a, two PCV2b, and six PCV2d isolates (Supplementary Table S1). The complete genome length of PCV2 in this study was 1768 bp (PCV2a) or 1767 bp (PCV2b and PCV2d). The 11 PCV2 strains from Jeju were genotypically cognate, having 99.2–99.7%, 99.4%, and 98.9–99.9% sequence identity within a corresponding genotype and shared 98.5–98.7%, 99.5–99.6%, and 99.0–99.9% identity with PCV2a, PCV2b, and PCV2d reference strains, respectively (Table 3). The percent identities of ORF1 and ORF2 of the Jeju isolates to one another and to the reference strains are summarized in Supplementary Table S2.
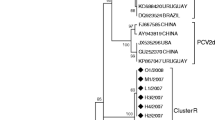
Aligning the genomes of all PCV2 sequences revealed marked variation between the genotypes but clear similarities within the genotypes (Fig. 1). However, one PCV2d strain, KNU-1931, showed significant sequence diversity in the ORF1 gene, specifically in the first 300 nucleotides, compared with those of other PCV2d strains. This region of KNU-1931 had significant sequence similarity to Korean and Chinese PCV2b strains, suggesting a potential natural recombination event. Phylogenetic analysis of the complete genome and ORF2 clearly defined the PCV2 strains into five genotype clusters, and all Jeju isolates were classified according to the respective genotypes (Fig. 2). However, the ORF1-gene-based phylogenetic analysis showed distinct tree topology and unique branches within the PCV2d cluster (Fig. 2C). Interestingly, the KNU-1931 strain was grouped with Korean and Chinese PCV2b strains (KU-1202-like), indicating the emergence of a novel recombinant strain (Fig. 2C).

Schematic diagram of a multiple alignment of the PCV2 genome relative to the consensus sequence derived from 74 global PCV2 strains, produced using Geneious software version 10.2.4. Genotypes of PCV2 are color-coded: PCV2a (blue), PCV2b (orange), PCV2c (purple), PCV2d (green), and PCV2e (pink). The Jeju PCV2 isolates identified during 2019 and 2020 in this study are indicated by an asterisk (*). The top illustration depicts the genomic regions, with green arrows symbolizing the identified ORFs. Lightly shaded regions show similarity to the consensus nucleotide sequence, and the vertical black bars represent variations from the consensus sequence. Thin horizontal dashed lines indicate deleted nucleotides.

Phylogenetic analysis based on the nucleotide sequences of the full-length genome (A), ORF2 (B), and ORF1 (C). The complete genome or corresponding gene sequences of a PCV1 strain were included as an outgroup. Multiple sequence alignments were performed using ClustalX, and phylogenetic trees were constructed from the aligned nucleotide sequences using the neighbor-joining method. The numbers at each branch indicate bootstrap values greater than 50% based on 1000 replicates. The names of the strains along with the country and year of isolation, GenBank accession numbers, and genotypes are shown. Red circles indicate the PCV2 strains identified in this study; a blue circle indicates the recombinant PCV2d strain identified in this study; a blue square indicates a major parental strain (PCV2d) identified in 2016; a red square indicates the minor parental strain (PCV2b) identified in 2012; open circles indicate the recombinant PCV2d strains identified in 2016. An ORF1-based KU-1202-like phylogenetic cluster containing KNU-1931 is shaded in yellow. Scale bars indicate nucleotide substitutions per site.
Genetic recombination analysis was performed using the RDP4 package to compare the KNU-1931 isolate and Korean strains to confirm the recombination event. Eight methods in the RDP4 platform (RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq, and LARD) were utilized to identify recombination events and breakpoints; the results are summarized in Table 4. All eight modules indicated that KNU-1931 emerged as a consequence of recombination, with a high degree of statistical support (average p-value, 9.004 × 10−5). These results defined KU-1602 (PCV2d genotype) and KU-1202 (PCV2b genotype) as the major and minor parental sequences, respectively (Fig. 3A). Furthermore, a similarity plot also indicated that KNU-1931 originated through natural intergenotypic recombination between PCV2b (KU-1202) and PCV2d (KU-1602) strains circulating in South Korea (Fig. 3B). Two putative recombination breakpoints were detected at nt 1746 and 299, which correspond to nt 4 in the origin of replication (ori) and nt 249 in ORF1, respectively. These data pointed to the introduction of the region from ori to the 5′ end of ORF1 of the parental KU-1202 strain into the backbone of the parental KU-1602 strain. In addition, the minor parental region (nt 1746–1767 and 1–299) of KNU-1931 showed higher sequence similarity to KU-1202 (99.3% identity) than to KU-1602 (95.6% identity), whereas the major parental region (nt 300–1745) of KNU-1931 showed greater sequence similarity to KU-1602 (99.6% identity) than to KU-1202 (95.9% identity).

Recombination analysis of KNU-1931. (A) Recombination detection. The x-axis indicates the genomic position, and the y-axis represents the pairwise identity between KNU-1931 and KU-1602; KNU-1931 and KNU-1202; or KU-1602 and KU-1202, illustrated using green, purple, and yellow lines, respectively. The beginning and end of the recombinant region are shaded red and labeled with position numbers. (B) Similarity plot analysis of KNU-1931 with KU-1602 (green) and KU-1202 (purple). The similarity plot was generated between KNU-1931 (query) and KU-1602 or KU-1202 using SimPlot v.3.5.1. with the two-parameter Kimura distance model and a window size of 200 bp and a step size of 20 bp. The x- and y-axes of the graph represent the nucleotide position (bp) and the percent nucleotide similarity, respectively. A yellow shaded area indicates the recombination region detected at nt 1746–1767 and 1–299, which encompass parts of the origin of replication (ori) and ORF1. (C) Phylogenetic trees of the major and minor parental regions of KNU-1931. The major parental region of KNU-1931 was closely related to the corresponding region of KU-1602, whereas the minor parental region was most closely related to the corresponding region of KU-1202.
Lastly, additional evidence of recombination was provided by statistically incongruent phylogenetic trees constructed using the major and minor parental regions, as well as base-by-base comparisons at genetic marker positions. KNU-1931 clustered with KU-1602 (PCV2d) in the phylogenetic tree of the major parental region, whereas its minor parental portion was more closely related to that of KU-1202 (PCV2b) (Fig. 3C). The base-by-base comparisons indicated that the nucleotide sequence of KNU-1931 was identical to that of KU-1202 within the breakpoints but was more similar to KU-1602 beyond the breakpoints (Fig. 4).

Base-by-base nucleotide comparisons of the recombination fragment of KNU-1931 (blue) and potential minor (KU-1202; purple) and major (KU-1602; red) parents. The Jeju PCV2d strains identified in this study are indicated by an asterisk (*). The recombination areas are shaded in yellow, and the solid boxes indicate the position of potential breakpoints.
Discussion
PCV2 causes severe financial losses in the global swine industry and needs to be controlled to improve production performance. Since its discovery, PCV2 has exhibited extraordinary genetic diversity, and it comprises five distinct genotypes in the current classification system. PCV2 has remained problematic and has continued to evolve in Korean pig populations since it was first reported in 1999 [16]. Similar to findings in other PCV2-endemic pig-raising countries, retrospective studies have revealed the existence of two genotype shifts, the first in 2002 from PCV2a to PCV2b and the second in 2012 from PCV2b to PCV2d, and that PCV2a, PCV2b, and PCV2d have co-circulated in Korean pig herds [17, 19, 26]. Although single PCV2d infection was a common occurrence in mainland South Korea as well as Jeju Province, multiple genotypes of PCV2 have coexisted in the same swine herd, resulting in dual or triple infections [19]. In the present study, we confirmed that three genotypes of PCV2 comingle and that PCV2d is the most prevalent genotype in Jeju Province of South Korea. Previously, more than 50% (8/14) of PCV2-positive pig farms in Jeju Province showed single infections with the PCV2d genotype [19]. However, all PCV2-positive cases in this study were coinfections with PCV2d and other genotypes in individual pigs, and single infections with one PCV2 genotype were found on Jeju pig farms. This result might reflect the smaller number (n = 8) of farms tested in the current study compared to a prior study (n = 14). Nevertheless, our data indeed indicate that minor genotypes of PCV2 still co-circulate at the farm level, which could not only contribute to altered pathogenicity of PCV2 but also facilitate viral recombination.
Genetic changes in viruses, including mutation and recombination, are the central driving forces of their evolution. PCV2 exhibits high rates of both nucleotide substitution and recombination [27, 28]. In particular, frequent intergenotypic and intragenotypic recombination play an important role in rapid PCV2 evolution and affect viral classification [13, 29,30,31,32,33,34,35]. Our extensive recombination analysis revealed that strain KNU-1931, identified in this study, is an intergenotypic recombinant resulting from the incorporation of a segment of KU-1202 (PCV2b) into the backbone of KU-1602 (PCV2d). The present analysis identified two potential recombination breakpoints at nt 1746 and 299, which are located in the distal region of ori and the proximal region of ORF1. The PCV genome primarily consists of two large ORFs, ORF1 and ORF2, located on the positive and complementary strand, respectively; the former is considered to be a hotspot of intergenotypic recombination, while the latter is regarded as a favorable region of intragenotypic recombination [18, 28, 29, 35,36,37]. Since ori is a relatively conserved region of the PCV2 genome, the possible breakpoint detected by RDP in this study might extend to this site. Given this probability, the ORF1 gene of a variant KNU-1931 strain would be a realistic target for natural intergenotypic recombination between the PCV2b and PCV2d genotypes simultaneously circulating in South Korea.
In addition, we identified KU-1202 (PCV2b) and KU-1602 (PCV2d) as the minor and major parental strains of the KNU-1931 recombinant, and these were isolated in different provinces of mainland South Korea in 2012 and 2016, respectively. Considering this geographic and temporal evidence, a natural intergenotypic recombination event between PCV2b and PCV2d might have occurred around 2016 on the mainland rather than on Jeju Island. Further investigation using Korean PCV2 sequences from the GenBank database confirmed the presence of two homologous recombinants from 2016, KU-1606 and KU-1607, that shared 99.8% nucleotide sequence identity with KNU-1931 in the present study (Fig. 1) and formed a monophyletic cluster with KNU-1931 in the PCV2d genotype (Fig. 2). These results suggest that the strain produced by the inter-genotypic recombination between PCV2b and PCV2d likely emerged in 2016 and then spread nationwide. Because the importation of live pigs or pork products from the mainland to Jeju Island is prohibited, we consider it likely that such a recombinant variant was introduced into Jeju pig herds by non-pig transmission sources, including traffic and humans, from the mainland.
Although vaccination is one of the effective strategies for control of PCV2 infection, global epidemics of PCV2d might be connected with cases of vaccine failure [38,39,40]. Indeed, despite vaccination having taken place on all eight farms examined in this study, the animals suffered from clinical PCVAD under field conditions. Dual heterologous infection with PCV2a and PCV2b has been shown to induce more-severe PCVAD than single infections [41]. Thus, coinfection with different PCV2 genotypes, including the recombinant PCV2d variant, appears to be associated with PCVAD in pigs with vaccine-induced immunity. Since an intergenotypic recombination event could allow the virus to undergo large-scale genomic changes resulting in novel variants with unusual traits or phenotypes, including modified pathogenicity, we cannot exclude the possibility that an emerging recombinant variant might possess altered virulence and clinical manifestations in field circumstances. Therefore, further studies should be conducted to provide fundamental clues regarding the correlations between recombination and viral pathogenicity and to evaluate the efficacy of vaccines against the new recombinant variant. Considering the coexistence of multiple genotypes and the frequency of intergenotypic recombination, we also need to improve the diagnostic and sequencing assays that are in use. Since the identification of recombinant PCV2b strains with a partial ORF1 of PCV2a after the first genotype shift [18], this is the first report to describe the existence of new PCV2d variants resulting from natural intergenotypic recombination following the second genotype shift in South Korea. Recombination events are inevitable under conditions of different co-circulating PCV2 genotypes and will eventually cause antigenic modification, thereby triggering the emergence of immune-escape variants. Therefore, continuous monitoring and surveillance of PCV2 evolution are of paramount importance for preparing effective measures against the emergence of novel variants or genotypes.
References
Allan GM, McNeilly F, Kennedy S, Daft B, Clarke EG, Ellis JA, Haines DM, Meehan BM, Adair BM (1998) Isolation of porcine circovirus-like viruses from pigs with a wasting disease in the USA and Europe. J Vet Diagn Invest 10:3–10
Segalés J (2012) Porcine circovirus type 2 (PCV2) infections: clinical signs, pathology, and Laboratory diagnosis. Virus Res 164:10–19
Segalés J, Olvera A, Grau-Roma L, Charreyre C, Nauwynck H, Larsen L, Dupont K, McCullough K, Ellis J, Krakowka S, Mankertz A, Fredholm M, Fossum C, Timmusk S, Stockhofe-Zursiden N, Beattie V, Armstrong D, Grassland B, Baekbo P, Allan G (2008) PCV-2 genotype definition and nomenclature. Vet Rec 162:867–868
Ramamoorthy S, Meng XJ (2009) Porcine circoviruses: a minuscule yet paradox. Anim Health Res Rev 10:1–20
Cheung AK (2006) Rolling-circle replication of an animal circovirus genome in a theta-replicating bacterial plasmid in Escherichia coli. J Virol 80:8686–8694
Khayat R, Brunn N, Speir JA, Hardham JM, Ankenbauer RG, Schneemann A, Johnson JE (2011) The 2.3-angstrom structure of porcine circovirus 2. J Virol 85:7856–7862
Olvera A, Cortey M, Segalés J (2007) Molecular evolution of porcine circovirus type 2 genomes: phylogeny and clonality. Virology 357:175–185
He J, Cao J, Zhou N, Jin Y, Wu J, Zhou J (2013) Identification and functional analysis of the novel ORF4 protein encoded by porcine circovirus type 2. J Virol 87:1420–1429
Liu J, Chen I, Kwang J (2005) Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J Virol 79:8262–8274
Davies B, Wang X, Dvorak CM, Marthaler D, Murtaugh MP (2016) Diagnostic phylogenetics reveals a new Porcine circovirus 2 cluster. Virus Res 217:32–37
Harmon KM, Gauger PC, Zhang J, Piñeyro PE, Dunn DD, Chriswell AJ (2015) Whole-genome sequences of novel porcine circovirus type 2 viruses detected in swine from Mexico and the United States. Genome Announc 3:e01315–e01315
Xiao CT, Halbur PG, Opriessnig T (2015) Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J Gen Virol 96:1830–1841
Guo LJ, Lu YH, Wei YW, Huang LP, Liu CM (2010) Porcine circovirus type 2 (PCV2): genetic variation and newly emerging genotypes in China. Virol J 7:273
Dupont K, Nielsen EO, Baekbo P, Larsen LE (2008) Genomic analysis of PCV2 isolates from danish archives and a current PMWS case-control study supports a shift in genotypes with time. Vet Microbiol 128:56–64
Franzo G, Cortey M, de Castro AM, Piovezan U, Szabo MP, Drigo M, Segalés J, Richtzenhain LJ (2015) Genetic characterisation of porcine circovirus type 2 (PCV2) strains from feral pigs in the Brazilian Pantanal: an opportunity to reconstruct the history of PCV2 evolution. Vet Microbiol 178:158–162
Lyoo YS, Kim JH, Park CK (1999) Identification of porcine circovirus with genetic variation from lymph nodes collected in pigs with PMWS. Korean J Vet Res 39:353–358
An DJ, Roh IS, Song DS, Park CK, Park BK (2007) Phylogenetic characterization of porcine circovirus type 2 in PMWS and PDNS Korean pigs between 1999 and 2006. Virus Res 129:115–122
Kim HK, Luo Y, Moon HJ, Park SJ, Keum HO, Rho S, Park BK (2009) Phylogenetic and recombination analysis of genomic sequences of PCV2 isolated in Korea. Virus Genes 39:352–358
Kwon T, Lee DU, Yoo SJ, Sang HJ, Shin JY, Lyoo YS (2017) Genotypic diversity of porcine circovirus type 2 (PCV2) and genotype shift to PCV2d in Korean pig population. Virus Res 228:24–29
Kim HR, Park YR, Lim DR, Park MJ, Park JY, Kim SH, Lee KK, Lyoo YS, Park CK (2017) Multiplex real-time polymerase chain reaction for the differential detection of porcine circovirus 2 and 3. J Virol Methods 250:11–16
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Martin DP, Murrell B, Khoosal A, Muhire B (2017) Detecting and analyzing genetic recombination using RDP4. Methods Mol Biol 1525:433–460
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Fulllength human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India with evidence of intersubtype recombination. J Virol 73:152–160
Kim D, Ha Y, Oh Y, Chae C (2011) Prevalence of porcine circovirus types 2a and b in pigs with and without post-weaning multi-systemic wasting syndrome. Vet J 188:115–117
Firth C, Charleston MA, Duffy S, Shapiro B, Holmes EC (2009) Insights into the evolutionary history of an emerging livestock pathogen: porcine circovirus 2. J Virol 83:12813–12821
Ramos N, Mirazo S, Castro G, Arbiza J (2013) Molecular analysis of Porcine Circovirus Type 2 strains from Uruguay: evidence for natural occurring recombination. Infect Genet Evol 19:23–31
Cai L, Han X, Hu D, Li X, Wang B, Ni J, Zhou Z, Yu X, Zhai X, Tian K (2012) A novel porcine circovirus type 2a strain, 10JS-2, with eleven-nucleotide insertions in the origin of genome replication. J Virol 86:7017
Franzo G, Cortey M, Segalés J, Hughes J, Drigo M (2016) Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol Phylogenet Evol 100:269–280
Jiang CG, Wang G, Tu YB, Liu YG, Wang SJ, Cai XH, An TQ (2017) Genetic analysis of porcine circovirus type 2 in China. Arch Virol 162:2715–2726
Liu J, Wei C, Dai A, Lin Z, Fan K, Fan J, Liu J, Luo M, Yang X (2018) Detection of PCV2e strains in Southeast China. PeerJ 6:e4476
Neira V, Ramos N, Tapia R, Arbiza J, Neira Carrillo A, Quezada M, Ruiz Á, Bucarey S (2017) Genetic analysis of porcine circovirus type 2 from pigs affected with PMWS in Chile reveals intergenotypic recombination. Virol J 14:191
Wang H, Gu J, Xing G, Qiu X, An S, Wang Y, Zhang C, Liu C, Gong W, Tu C, Su S, Zhou J (2019) Genetic diversity of porcine circovirus type 2 in China between 1999–2017. Transbound Emerg Dis 66:599–605
Wei C, Lin Z, Dai A, Chen H, Ma Y, Li N, Wu Y, Yang X, Luo M, Liu J (2019) Emergence of a novel recombinant porcine circovirus type 2 in China: PCV2c and PCV2d recombinant. Transbound Emerg Dis 66:2496–2506
Hesse R, Kerrigan M, Rowland RR (2008) Evidence for recombination between PCV2a and PCV2b in the field. Virus Res 132:201–207
Huang Y, Shao M, Xu X, Zhang X, Du Q, Zhao X, Zhang W, Lyu Y, Tong D (2013) Evidence for different patterns of natural inter-genotype recombination between two PCV2 parental strains in the field. Virus Res 175:7886
Eddicks M, Fux R, Szikora F, Eddicks L, Majzoub-Altweck M, Hermanns W, Sutter G, Palzer A, Banholzer E, Ritzmann M (2015) Detection of a new cluster of porcine circovirus type 2b strains in domestic pigs in Germany. Vet Microbiol 176:337–343
Salgado JM, Rodríguez-Solana R, Curiel JA, de Rivas B, Munoz R, Domínguez JM (2014) Bioproduction of 4-vinylphenol from corn cob alkaline hydrolyzate in two-phase extractive fermentation using free or immobilized recombinant E. coli expressing pad gene. Enzyme Microb Technol 58–59:22–28
Seo HW, Park C, Kang I, Choi K, Jeong J, Park SJ, Chae C (2014) Genetic and antigenic characterization of a newly emerging porcine circovirus type 2b mutant first isolated in cases of vaccine failure in Korea. Arch Virol 159:3107–3111
Harding JCS, Ellis JA, McIntosh KA, Krakowka S (2010) Dual heterologous porcine circovirus genogroup 2a/2b infection induces severe disease in germ-free pigs. Vet Microbiol 145:209–219
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF- 2018R1D1A1B07040334).
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF- 2018R1D1A1B07040334).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Handling Editor: Ana Cristina Bratanich.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jang, G., Yoo, H., Kim, Y. et al. Genetic and phylogenetic analysis of porcine circovirus type 2 on Jeju Island, South Korea, 2019–2020: evidence of a novel intergenotypic recombinant. Arch Virol 166, 1093–1102 (2021). https://doi.org/10.1007/s00705-020-04948-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04948-1