
Abstract
Disease outbreaks caused by turnip yellows virus (TuYV), a member of the genus Polerovirus, family Luteoviridae, regularly occur in canola and pulse crops throughout Australia. To understand the genetic diversity of TuYV for resistance breeding and management, genome sequences of 28 TuYV isolates from different hosts and locations were determined using high-throughput sequencing (HTS). We aimed to identify the parts of the genome that were most variable and clarify the taxonomy of viruses related to TuYV. Poleroviruses contain seven open reading frames (ORFs): ORF 0–2, 3a, and 3–5. Phylogenetic analysis based on the genome sequences, including isolates of TuYV and brassica yellows virus (BrYV) from the GenBank database, showed that most genetic variation among isolates occurred in ORF 5, followed by ORF 0 and ORF 3a. Phylogenetic analysis of ORF 5 revealed three TuYV groups; P5 group 1 and group 3 shared 45–49% amino acid sequence identity, and group 2 is a recombinant between the other two. Phylogenomic analysis of the concatenated ORFs showed that TuYV is paraphyletic with respect to BrYV, and together these taxa form a well-supported monophyletic group. Our results support the hypothesis that TuYV and BrYV belong to the same species and that the phylogenetic topologies of ORF 0, 3a and 5 are incongruent and may not be informative for species demarcation. A number of beet western yellow virus (BWYV)- and TuYV-associated RNAs (aRNA) were also identified by HTS for the first time in Australia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Turnip yellows virus (TuYV) was first identified in the United Kingdom as a European strain of beet western yellows virus (BWYV) based on biology and serology [19]. The International Committee on Taxonomy of Viruses (ICTV) later classified TuYV and BWYV as members of distinct species due to differences in their host range [50].
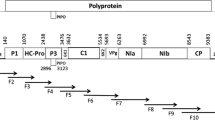
TuYV belongs to the genus Polerovirus (family Luteoviridae), whose members have monopartite linear single-stranded RNA genomes encapsidated in an icosahedral shell [5, 11]. The RNA genome of TuYV contains seven open reading frames (ORFs). The 5’ ORFs 0, 1 and 2 are expressed by translation of genomic RNA and encode the proteins P0 and P1 and the P1-P2 fusion protein, which are associated with pathogenicity, host range, silencing suppression and replication [6, 31, 49, 58, 74, 75, 82]. The 3’ portion of the genome is separated from ORFs 0, 1 and 2 by an intergenic region (IR) and contains ORFs 3, 3a, 4 and 5, which encode the proteins P3, P3a, P4 and P3-P5 via translation of subgenomic RNA [49, 71]. P3 is the coat protein (CP), P3a and P4 are movement proteins (MP), and the P3-P5 readthrough domain (RTD) is required for virus circulation, accumulation and persistence in the aphid vector [7, 9, 49, 60, 65, 71, 82].
Members of each Polerovirus species are transmitted by one or more aphid species in a persistent, circulative manner, with varying degrees of efficiency [64, 72]. The most efficient vector for TuYV is the green peach aphid Myzus persicae, with reported transmission rates of over 90% [64]. Other aphid species, such as Brevicoryne brassicae, Aphis gossypii and Macrosiphum euphorbiae, vector TuYV with lower rates of transmission (less than 10%) [64, 72].
TuYV infects a number of crops and weeds belonging to a wide range of plant families, including Brassicaceae, Fabaceae, Amaranthaceae and Asteraceae [73]. The broad host range of TuYV, which includes both summer and winter crops as well as opportunistic weeds, increases the potential reservoir of virus inoculum, providing a “green bridge” for both viruses and their aphid vectors [25].
The diversity of hosts and potential vectors of TuYV have contributed to its worldwide distribution. TuYV has been reported throughout Europe [73] and in Iran [67], China [77], South Africa [53], Colombia [62], Saudi Arabia (unpublished; GenBank accession no. LT844559), Egypt [1], Morocco [1] and Australia [14].
New strains and species of viruses related to TuYV and BWYV have recently been identified based on i) genome sequence analysis and ii) the ICTV species demarcation rule stating that viruses of the family Luteoviridae are members of distinct species if the amino acid sequence of any gene product differs by more than 10%. For example, brassica yellows virus (BrYV) was separated from TuYV based on differences in ORF 0 and ORF 5 [41, 79, 81]. Faba bean polerovirus 1 (FBPV-1), identified in winter legume crops (pulses) in Australia, is the result of recombination between an unknown virus and TuYV [24], and beet leaf yellowing virus (BLYV), identified in Japan, differs from BWYV, predominantly in ORF 0 and ORF 1 [80].
Full genome sequencing of poleroviruses infecting Capsicum annuum, such as pepper vein yellows virus (PeVYV) from Japan, China, Australia, Spain and Greece, and pepper yellow leaf curl virus (PYLCV) from Israel, showed that the sequence variation between these viruses exceeds 10% in ORF 0, 3a and 5 [17, 42, 43, 45]. Fiallo-Olivé et al. [22] showed that the genetic variation in pepper poleroviruses is complex due to recombination events and proposed that these viruses should be described as numbered species (PeVYV 1-6) in order of discovery rather than being given new species names.
TuYV epidemics occur regularly in Australia, for example, in 2012 in chickpeas (Cicer arietinum) from northern New South Wales [76], in 2014 and 2018 in canola (Brassica napus) from south Western Australia [15] and Esperance in Western Australia [12], in 2014 in canola from Riverton, South Australia and Victoria, and more recently, in 2019 in canola from southern New South Wales (Joop Van Leur, unpublished). In Europe, TuYV is a constant threat to production, with regular epidemics and significant yield losses [3, 54, 66].
To understand the genetic diversity of TuYV, we used high-throughput sequencing (HTS) to determine the full genome sequences of TuYV from pulses, canola, and weeds in Australia. We aimed to i) identify the parts of the genome of TuYV that are most variable, ii) determine whether there is recombination in populations of TuYV from Australia, and iii) better understand the taxonomy of viruses related to TuYV. Knowledge of the genomic diversity of viruses can assist in crop management, diagnostics, and development of disease resistance.
Materials and methods
Collection of plant material
Leaves and stems of plants were collected between 2013 and 2018 during surveys of plant viruses from various locations around Australia (Table 1 and Supplementary Fig. 1). Selected TuYV isolates were freeze-dried and stored in the Queensland Department of Agriculture and Fisheries (DAF) virus collection. Overseas isolates were collected between 2004 and 2014 from Algiers in Algeria, Yunnan Province in China, the Bekaa Valley in Lebanon, Marchouch in Morocco, Hudiba in Sudan, Bizerte in Tunisia, and Tashkent and Kashkadarya in Uzbekistan (Table 3).
Tissue blot immunoassays and ELISA
Virus-positive samples were identified by tissue blot immunoassay (TBIA); cross sectioned stems of each plant were blotted onto nitrocellulose membranes (Amersham™ Proton™ 0.45 µm NC, Merck, New Jersey, USA) as described by Makkouk and Comeau [46]. TBIAs were processed as described by Filardo and Sharman [23]. The primary antibody used to detect poleroviruses, including TuYV, was the broad-spectrum luteovirid monoclonal antibody BLRV-5G4 [2, 34].
RT-PCR and sequencing
Total nucleic acid and total RNA were isolated using either a BioSprint 15 workstation with a BioSprint 15 Plant DNA Kit (QIAGEN, catalogue no. 941517) without the use of RNase A or with a QIAGEN RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) with a modified lysis buffer [44], as per manufacturer’s instructions.
Initially, TBIA-positive samples were amplified by RT-PCR using the primer pair BWYV3969F/AS5 (Table S1), which detects TuYV and BWYV but does not distinguish between the two viruses [24]. cDNA was synthesised using SuperScript III reverse transcriptase (Thermo Fisher Scientific, Waltham, USA) or an ImProm-II™ Reverse Transcription System (Promega, Wisconsin, USA) as per instructions, using primer AS3 or AS5 (Table S1). PCR conditions were as described by Sharman et al. [68], using the primers BWYV3969F (Table S1) and AS5 under the following cycling conditions: initial denaturation at 95 °C for 1 min, then 35 cycles of 95 °C for 15 s, 62 °C for 20 s, 56 °C for 10 s, and 72 °C for 40 s, followed by a final extension step at 72 °C for 3 min.
RT-PCR assays to distinguish the different TuYV ORF 5 domains used a one-step RT-PCR QIAGEN kit as per manufacturer’s instructions and the primer pairs TuYV-4841F/TuYV-5328R for TuYV group 1, TuYV-4841F/BrYV-5476R for group 2, and BrYV-4680F/BrYV-5476R for group 3 (Table S1). The three primer sets were used individually or together in a multiplex RT-PCR format, with a 0.2 µM final concentration of each primer under the following cycling conditions: 50 °C for 30 min and 95 °C for 15 min, followed by 35 cycles of 94 °C 30 s, 56 °C for 30 s, and 72 °C for 50 s, followed by a final extension step at 72 °C for 10 min. The RT-PCR fragments for TuYV groups were distinguished by size on a 1% agarose gel – group 1 was 487 bp, group 2 was 637 bp, and group 3 was 772 bp.
RT-PCR assays to amplify the intergenic region (IR) and ORF 3a had the same reaction conditions described above with primers AS5 and primer TuYV-3299F (amplifies IR-A) or TuYV-3394F (IR-B) (Table S1), using cycling conditions described by Congdon et al. [13].
The degenerate primers aRNAv-1120F and aRNAv-1510R were used in RT-PCR to amplify virus-associated RNAs, such as BWYV ST9 aRNA and TuYV sat1 aRNA, using the conditions described above with an annealing temperature of 60 °C. The specific primers ST9-1198F and ST9-1818 were used to amplify BWYV ST9 aRNA.
PCR products were sequenced directly (Macrogen Inc. Seoul, Korea) and analysed using Geneious 9.0 (Biomatters).
High-throughput sequencing
Total RNA was extracted from TuYV isolates (Table 1) using a Spectrum Plant Total RNA Kit (Merck, New Jersey, USA) as described by Filardo et al. [24] or using a QIAGEN RNeasy Plant Mini Kit with a modified lysis buffer [44]. For TuYV isolates from Victoria, libraries were prepared using an NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, USA), quantified using a 2200 TapeStation system (Agilent Technologies, California, USA) and sequenced using an Illumina MiSeq system as described by Kinoti et al. [36]. For the other TuYV isolates, paired-end libraries (150 bp) were prepared and sequenced using an Illumina MiSeq or HiSeq 2500 system by the Australian Genome Research Facility (AGRF, Melbourne, Australia). Adaptor and primer sequences were removed from the reads in Geneious 9, using the BBDuK plugin (part of the BBTools package) [10].
Trimming for quality, pairing of reads, and contig assembly were done as described by Filardo et al. [24] or trimmed using Trim Galore v 0.4.4 [37] with the minimum sequence length set to 50 bp and the stringency set to 1 bp. De novo assembly was performed using the SPAdes Genome Assembler [4] or CLC Genomics Workbench 6.5 (CLCGW) (QIAGEN, Hilden, Germany), both with default settings. Contigs were sorted by length and subjected to a BLAST search [32]. Contigs and BLAST results were analysed further in Geneious 9 (Biomatters, New Zealand). Mapping of contigs and reads to a reference genome, TuYV-FL1 (NC_003743), was performed with the following settings: minimum overlap, 10%; minimum overlap identity, 80%; allow gaps, 10%; fine tuning set to iterate up to 10 times. Reference mapping of reads to final full-length contigs was done in Geneious 9 or using Bowtie 2 v 2.3.4.2 [40] with default settings.
Three representative full genome sequences of TuYV obtained by HTS were confirmed by Sanger sequencing; TuYV-Br12 (ORF 5 group 1), TuYV-5509 (ORF 5 group 2), and TuYV-5510 (ORF 5 group 3). RT-PCR was performed using a SuperScript™ III One-Step RT-PCR Kit (Invitrogen, California, USA) in 25-µl reactions according to the manufacturer’s instructions, using the following cycling conditions: 48 °C for 45 min and 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 55 or 58 °C for 30 s, and 72 °C for 50 s, followed by 72 °C for 10 min. The primer sets used were TuYVSeg1F/R, TuYVSeg2F/R, TuYVSeg3F/R, TuYVSeg4F/R, TuYVSeg5_5F/1R, TuYV3564F/4372R, TuYV4372F/5201R and TuYV5183F/6097R (Table S1). PCR products were purified using a QIAquick PCR Purification Kit (QIAGEN) and sequenced directly by AGRF. Open reading frames (ORFs) were predicted and annotated using Geneious 9.
Phylogenetic and phylogenomic analysis
Phylogenetic analysis was done for single genes and the predicted amino acid sequences of the proteins encoded by the six ORFs from 28 TuYV isolates from this study as well as previously sequenced TuYV isolates from Australia and other poleroviruses (Table 1) based on alignments made using ClustalW with a BLOSUM cost matrix in Geneious 10. Phylogenetic analysis for each ORF was conducted in MEGA 7 [39] using the maximum-likelihood method based on the JTT matrix-based model [33] and 500 bootstrap replicates.
Phylogenomic analysis was done using the entire coding region of the genome. The aligned ORFs were concatenated and searched for the most likely tree in IQTree v.1.7 beta [55] with a model test for each partition (command: -spp -m TEST), 10,000 ultrafast bootstrap replicates [29], an approximate likelihood ratio test with 10,000 replicates [27], and genealogical concordance factors calculated from each locus [51].
Recombination analysis, pairwise identity, and frequency distribution plots
We visualised putative recombination events in the TuYV genome based on genetic distance between genome sequences in SplitsTree4 [30]. We tested for recombination in ORF 5 of TuYV group 2 isolates; aligned sequences of TuYV-FL1, BrYV-CR, and TuYV-5509 were examined using RDP4 [47]. Pairwise identities of 60 aligned whole-genome nucleotide sequences were displayed as a matrix using sequence demarcation tool (SDT) v. 1.2 [52].
Frequency distribution plots were generated by aligning sequences of all TuYV and BrYV isolates, cucurbit aphid borne yellows virus (CABYV), phasey bean mild yellows virus (PBMYV), siratro latent polerovirus (SLPV), faba bean polerovirus 1 (FBPV-1), and beet western yellows virus (BWYV). For this purpose, full genome nucleotide sequences or the deduced amino acid sequences of the RdRp or CP were used, and a pairwise distance chart was generated using the P-distance model in MEGA7 [39]. Results were converted to percentage pairwise identity. The frequency of each pairwise value was determined and plotted against percentage pairwise identity.
Results
HTS analysis of TuYV in Australia
Between 2016 and 2018, 25 TuYV-positive samples were collected from various hosts and locations across Australia, and their genomes were sequenced and assembled (Table 1, Supplementary Fig. S1). In total, 3,376,590-19,797,660 reads were obtained for each sample, reduced to 3,353,970-19,323,967 after trimming for quality (Supplementary Table S2). The number of contigs produced after de novo assembly across the 25 samples was 92-15,212, with the TuYV contigs with lengths of 499-5,701 nt. The final genome sequence lengths after mapping and contig analysis ranged from 5,474 to 5,786 nt, with the average final coverage ranging from 18.2 to 59,630 in depth (Supplementary Table S2). From the 25 samples used in this study, some contained mixed infections so that a total of 28 TuYV genomes were identified. Genomes of three representative TuYV isolates, TuYV-Br12, TuYV-5509 and TuYV-5510, were re-sequenced by the Sanger method, verifying the HTS results with 99.98–100% nt sequence identity.
Phylogenetic analysis
The aligned open reading frames (ORFs 0-5) of the 28 TuYV isolates from this study and TuYV isolates and related poleroviruses from GenBank (Table 1) showed that the genomes of TuYV were diverse, with most genetic variation in ORF 5 followed by ORF 0 and ORF 3a (Fig. 1).

Phylograms from a maximum likelihood search of amino acid sequence alignments of TuYV, BrYV and other closely related members of the genus Polerovirus constructed in MEGA. Phylograms based on P5 (ORF5), P0 (ORF 0) and P3a (ORF 3a). Accession numbers are listed in Table 1. Colouring of the TuYV, BrYV and BWYV isolates relates to different monophyletic groups based on P5. The assigned P5 colour is carried through into the P0 and P3a phylograms, to aid in the visualisation of the diversity found within the TuYV genomes
ORF 5
Phylogenetic analysis of P5 (ORF 5) separated TuYV into three groups (Fig. 1). Group 1 contained the type member TuYV-FL1 and 14 TuYV isolates with 89–100% aa sequence identity (Fig. 1). Group 2 contained 20 TuYV Australian isolates and BrYV-CS, sharing 94-100% aa sequence identity. Group 3 aligned with the newly described BrYV from China and contained six TuYV Australian isolates that had 92–100% aa sequence identity (Fig. 1).
Groups 1 and 3 were the most genetically divergent between groups, sharing 45–49% aa sequence identity in P5 (Fig. 1; Table 2). Group 2 shared 60–64% aa sequence identity with group 1 and 77–84% aa sequence identity with group 3 (Fig. 1; Table 2).
Diversity within the P5 protein was also observed for the BWYV isolates included in the phylogenetic analysis (Table 1). The isolates BWYV-BJA and BWYV-BJB shared 23% aa sequence identity with the BWYV-USA type member in P5 (Fig. 1; Supplementary Table 4).
ORF 0
The TuYV and BrYV isolates share 77–100% amino acid sequence identity in P0 (Fig. 1; Table 2). Despite the amino acid sequence variation, phylogenetic analysis showed all isolates of TuYV and BrYV being in the same monophyletic group, sharing a common ancestor for this region of the genome.
ORFs 3a, 1, 2, 3 and 4
The small non-AUG-initiated P3a protein of TuYV occurred in two monophyletic groups that shared 69–73% aa sequence identity with each other (Fig. 1). Group 1, based on amino acid sequences of P5, all had a similar copy of P3a (ORF 3a-1) (Fig. 1). P5 groups 2 and 3 contained either P3a-1 or P3a-2 (Fig. 1).
The P3a proteins of the BWYV isolates and BLYV shared 87–91% aa sequence identity with all other BWYV isolates except BWYV-USA. The type member, BWYV-USA, shared 67–73% aa sequence identity with the other BWYV isolates and BLYV (Fig. 1).
Analysis of the amino acid sequences of P1, P2, P3 and P4 showed that these regions were more conserved than ORFs 5, 0, and 3a. The TuYV and BrYV isolates formed monophyletic groups for each of the following open reading frames with the following amino acid sequence identity values: P1, 83–100 %; P2, 90–100%; P3, 90–100%; P4, 86–100% (Table 2; Supplementary Figs. S2 and S3).
Recombination in ORF 5
To check for potential recombination events and to identify the breakpoint between TuYV isolates from different assigned ORF 5 groups, a representative sequence from each group (TuYV-FL1, BrYV-CR, and TuYV-5509) was aligned and examined using the recombination program RDP4. A putative recombinant event for TuYV-5509, with TuYV-FL1 and BrYV-CR, was strongly supported using the RDP, GENECONV, Bootscan, Maxchi, Chimaera, and 3Seq methods, all with p-values less than 1 × 10-16 (Fig. 2b). The predicted breakpoint is located approximately 786 nt downstream of the start of ORF 5 of TuYV-5509. The first 786 nt of TuYV-5509 ORF 5 align closely with TuYV-FL1 (93% aa sequence identity), while TuYV-FL1 and BrYV-CR share only 69% aa sequence identity in this region. The 3’ half of TuYV-5509 ORF 5 (787 nt into ORF 5 to the end of the reading frame, ~ 898 nt in length), shares 91% aa sequence identity with BrYV-CR and only 43% aa sequence identity with TuYV-FL-1. The same pattern of recombination in ORF 5 was observed in all TuYV group 2 isolates (data not shown).

a Schematic representation of TuYV group genomes with the seven ORFs (ORF 0–5 & 3a). The dark grey ORFs depict the proteins that are monophyletic. The ORF 5 region is coloured purple for group 1 and green for group 3. For group 2 ORF 5, the recombinant region is depicted, with group 1 (purple) and group 3 (green). The small ORF 3a protein is depicted in yellow–yellow with no stripe represents P3a-1 only and yellow with stripe represents the group containing either P3a-1 or P3a-2. b Results of the RDP recombination analysis for TuYV-5509 (ORF 5 group 2). Lines indicate the percentage of similarity per alignment and the pink area indicates the recombinant part of the sequence. In this analysis TuYV-FL1 and BrYV-CR contained P3a-1 and TuYV-5509 contained P3a-2. c Frequency distribution plot based on whole genome nucleotide identities of 47 TuYV and BrYV isolates, BWYV, CABYV, PBMYV, SLPV and FBPV-1. Sequence identities between distinct viruses had a mean value of 58.5 % and a standard deviation of 6.7 %, while identities between strains of individual viruses (TuYV/BrYV) showed a mean of 90.5 % ± 4.0 %
Recombination analysis and phylogenetic analysis showed that the genomes of ORF 5 TuYV group 2 isolates contain part of ORF 5 from TuYV group 1 and part of ORF 5 from TuYV group 3 (BrYV) as well as either ORF 3a version 1 or version 2, as shown in the schematic representation in Fig. 2a.
Host range of TuYV groups
The TuYV isolates from all three groups were found in a range of pulse and canola crops, as well as weeds, throughout Australia (Table 1). During surveys conducted along the east coast of Australia (QLD, NSW, and VIC) between 2015 and 2019, plants that were positive by PCR (BWYV-3969F + AS5) for TuYV were retested in a multiplex RT-PCR that distinguished the different P5 groups. Table 3 shows the range of host species for each TuYV group recorded to date in Australia.
A small range of TuYV-positive samples collected between 2013 and 2014 from various countries were also retested using the P5 multiplex PCR. These results show that the different TuYV groups are not confined to Australia (Table 3).
Phylogenomic analysis
Despite the amino acid sequence variation observed in P5, P0, and P3a, phylogenomic analysis showed that TuYV and BrYV share a common ancestor, and together, these isolates form a well-supported monophyletic group (Fig. 3). There was phylogenetic support for BrYV (1.0 aLRT, 100% UltraFast Bootstrap), which formed a nested, monophyletic group in the concatenated analyses; however, this clade was not reproduced in the gene trees of the ORFs (genealogic concordance factor = 0).

Phylogram obtained from a maximum likelihood search in IQTree v1.7, based on aligned genomes with a model test for each ORF. aRLT values (≥ 0.9) and ultrafast bootstrap values (≥ 95%) from 10,000 replicates, and genealogical concordance values from each partition above nodes
Our SplitsTree analysis suggested that recombination events had occurred between TuYV and BrYV (ORF 5; Fig. 2a and b) and that these viruses are closely related by genetic distance (Fig. 3). There was more intraspecific diversity across the genomes of BWYV than between the genomes of TuYV and BrYV based on genetic distance (Fig. 3).
Pairwise identity and frequency distribution
The full-genome pairwise identity matrix showed a genetic continuum among the TuYV and BrYV isolates, with nucleotide sequence identity ranging from 82 to 99 % (Fig. 4). There was no distinct division within the genetic continuum in which a species separation could be inferred, supporting a monophyletic hypothesis for TuYV and BrYV (Fig. 3).

Colour matrix of pairwise nucleotide sequence identity inferred from alignments of whole genomes of TuYV, BrYV and other closely related poleroviruses
Frequency distribution plots can be used to distinguish virus species and aid in species demarcation when paired sequence identity values are plotted against frequency, resulting in a bimodal distribution of sequence identities [52, 69]. A whole-genome frequency distribution plot with all TuYV/BrYV isolates, BWYV-USA, CABYV, PBMYV, SLPV and FBPV-1, showed that sequence identity between distinct polerovirus members (first bimodal curve) ranged from 51 to 71 % (average, 54.0 ± 6.7 %) (Fig. 2c). The TuYV/BrYV isolates formed a second bimodal curve with a continuum range of 83 to 100 % (average, 90.5 ± 4.0 %), showing that all TuYV isolates group together as strains with a wide distribution of identity values.
To determine a region within the TuYV/BrYV genome that can be used for future diagnostics and species demarcation and reflect the phylogenomic, pairwise identity, and full-genome frequency distribution plot results, we plotted the frequency distribution of amino acid sequences of the conserved RdRp (ORF 2) and CP (ORF 3) regions (Supplementary Fig. 4). These regions show a bimodal distribution of sequence identity values with distinct species ranging from 60 to 67% (average, 64.1 ± 1.6 %) for the RdRp region and from 65 to 74% (average, 68.8 ± 0.9) for the CP. Identities between strains of TuYV/BrYV ranged from 91 to 100% (average, 95.8 ± 1.9 %) and from 89 to 100 % (average, 94.5 ± 2.2 %) for RdRp and CP, respectively (Supplementary Fig. S4). The only exception to the bimodal pattern of species and strains was the BWYV-USA isolate, which grouped with TuYV/BrYV isolates for the CP, due to the high degree of similarity (range, 87–92% identity) in this region.
Identification of virus-associated RNAs
Analysis of HTS data identified eight virus-associated RNA (aRNA) sequences in TuYV-infected canola and one in wild mustard (Sinapis arvensis) (Table 4). Six of the aRNAs (5248, 5512a, C20A, C21A, C2019, 5414) shared 97–100% nt sequence identity and were identified as BWYV aRNA, NC_004045 (93% nt sequence identity to BWYV ST9 aRNA; [11]) (Table 4). Four aRNAs, 5512b from NSW, 5514 from SA, and C20A8 and C2016-17 from VIC, all from canola, shared 93% nt sequence identity with the newly identified TuYV sat1 aRNA (MN164515; unpublished) and only 79–80% nt sequence identity with BWYV ST9 aRNA (Table 4; Fig. 5). We have tentatively named the new TuYV aRNA isolates “TuYV Oz aRNA”.

Maximum likelihood phylogenetic tree based on nucleotide sequence alignments of aRNAs identified in Australia.
Discussion
In this study, we provide evidence that the P5 readthrough domain, P0, the small movement P3a protein, and, to a lesser extent, P1, are genetically diverse in isolates of TuYV/BrYV and BWYV. Genetic variation in these ORFs has been reported for TuYV isolates from England and continental Europe [3, 54] as well as for PeVYV-like isolates [22]. This suggests that polerovirus genomes are more diverse within these regions than originally thought, and with increased whole-genome sequencing, more diversity is likely to be found.
We showed that ORF 5 is a genetically variable region with evidence of recombination between two related isolates; the ORF 5 of group 2 TuYV isolates is a recombinant of group 1 and group 3 ORF 5. Recombination events in RNA viruses are well documented [28, 70, 78], and poleroviruses show that both intraspecific, homologous (TuYV and BrYV isolates) and interspecific, non-homologous (FBPV-1; [24]) recombination occurs.
We suggest that intraspecific variation in these genes is expected in a population, as these genes may be under selection from their hosts and vectors and accumulate changes faster than other parts of the genome. P5 is required for efficient aphid transmission and phloem retention of the virus and influences symptom development and virus accumulation [8, 9, 59, 60], P0 suppresses host resistance [5, 6, 58], and P3a is a movement protein – all of which would be associated with a compatible interaction [16, 26, 61].
Despite the genetic variation in ORFs 5, 0, and 3a, our phylogenomic analysis suggests that TuYV and BrYV are conspecific (i.e., belong to the same species). This was supported by a concatenated alignment of the entire genome in which TuYV and BrYV formed a monophyletic group.
The current ICTV species demarcation criteria for poleroviruses include differences in host range, failure to cross-protect, differences in serological reactions, and differences in the amino acid sequence of any gene product greater than 10% [35]. However, our analysis shows that the phylogenetic topologies of ORF 0, 3a, and 5 are incongruent, suggesting that comparison of these genes is not informative for species demarcation.
Our results suggest that it would be more feasible to classify TuYV isolates, and possibly other poleroviruses, according to whole-genome analysis. The whole-genome frequency distribution plot (Fig. 2c), which can also be used for determining species demarcation cutoffs [52, 69], suggests that TuYV and BrYV should be considered members of the same species if they share at least 83% nucleotide sequence identity. We therefore suggest that the species demarcation criteria for new poleroviruses would ideally include sharing less than 83% nt sequence identity in the whole coding region of the genome as well as any marked host range, vector, and serological differences.
For diagnostics purposes, whole genome sequences are not always attainable; therefore, species demarcation could also take into consideration conserved regions of the genome. For RNA viruses, the RdRp region, which is the only protein shared by all RNA viruses [38], and the coat protein are highly conserved within poleroviruses, and generally, conservation within these regions is indicative of membership in the same species [22, 48]. All TuYV/BrYV isolates share 91–100% aa sequence identity in the RdRp (P2) and 89–100% aa sequence identity in the CP (P3) (Supplementary Figs. S2, S3 and S4), reflecting the current species demarcation rules of less than 10% aa sequence difference in these proteins. We therefore suggest that members of the same polerovirus species should share more than 89% aa sequence identity in the RdRp and CP proteins. However, the intergenic region between the RdRp and CP is a known hotspot for recombination events [56]. Characterisation of the region spanning the RdRp and CP genes, for diagnostics and demarcation, would identify potential recombination events in these conserved regions. If a recombination event is suspected, this might indicate that the isolate belongs to a new species, and characterisation of the complete genome would be warranted.
There is preliminary evidence suggesting that genetic variation within TuYV ORF 0 may influence the host range of different TuYV isolates [3, 54]. However, the TuYV sequence data from Newbert [54] are not publicly available or published and therefore could not be included in this study. At this stage, there is no information about the biological significance of the variation in TuYV ORF 5. TuYV has a broad host range, and the complex genetic diversity among TuYV strains may be the reason for its success. RNA viruses typically exploit high mutation rates, with populations consisting of “swarms” of mutants and/or quasispecies to achieve resistance-breaking, changes in host range, persistence in the host, and long-term survival in nature [18, 28].
During HTS analysis, a number of associated RNA (aRNA) sequences were obtained, predominantly from canola. This is the first report of TuYV and BWYV aRNAs in Australia. Polerovirus-associated RNAs are ~2.8 kb in length with two major ORFs. They replicate independently but are dependent on viruses, such as BWYV, for encapsidation and probably for aphid transmission as well as systemic movement within plants [11, 21, 57, 63]. Six aRNAs identified in this study were found to be related to BWYV ST9 aRNAs. Coinfection with BWYV ST9-aRNA has been shown to increase BWYV virion yield and increase pathogenicity [20, 21, 57]. Four other aRNA isolates were found to be related to the newly identified TuYV aRNA sat1 from South Africa (MN164515; unpublished).
Currently, little is known about the role of associated RNAs in TuYV outbreaks. In this study, we identified two different aRNAs, both in hosts infected with TuYV - an associated RNA closely resembling those reported from BWYV (BWYV ST9 aRNA), despite not finding evidence of BWYV in Australia, and a TuYV aRNA. Interestingly, a canola plant collected in South Australia during a severe TuYV outbreak in 2014 – TuYV-5514b (as well as MK111 and MK113) contained an isolate of TuYV aRNA as well as all three TuYV P5 strains (confirmed by HTS and TuYV MP-PCR). Do associated RNAs play a role in TuYV outbreaks? Do TuYV strains form a complex that influences pathogenicity? These are future research questions that could lead to a better understanding of the epidemiology of TuYV outbreaks for this complex, genetically diverse virus.
Our study of TuYV diversity in Australia has raised questions about the currently used species demarcation criteria for poleroviruses based on aa sequence differences in gene products. As such, we have proposed a new method to better reflect the observed genetic diversity. This study also provides the knowledge and tools to investigate whether the diverse TuYV groupings identified actually have any biological differences, such as host range, vector specificity, or reaction with resistance genes, which may indicate that they are distinct strains or members of different species, which will require targeted control measures in affected crops.
References
Abraham AD, Varrelmann M, Vetten HJ (2008) Molecular evidence for the occurrence of two new luteoviruses in cool season food legumes in Northeast Africa. Afr J Biotechnol 7:414–420
Asaad NY, Kumari SG, Haj-Kassem AA, Shalaby A-BA, Al-Shaabi S, Malhotra RS (2009) Detection and characterization of Chickpea Chlorotic Stunt Virus in Syria. J Phytopathol 157:756–761
Asare-Bendiako E (2011) Brassicaceae: turnip yellows virus interactions. Doctoral dissertation, University of Warwick, Coventry. http://webcat.warwick.ac.uk/record=b2553527~S1
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Baumberger N, Tsai C-H, Lie M, Havecker E, Baulcombe David C (2007) The polerovirus silencing suppressor P0 targets ARGONAUTE proteins for degradation. Curr Biol 17:1609–1614
Bortolamiol D, Pazhouhandeh M, Marrocco K, Genschik P, Ziegler-Graff V (2007) The polerovirus F box protein P0 targets ARGONAUTE1 to suppress RNA silencing. Curr Biol 17:1615–1621
Brault V, van den Heuvel JF, Verbeek M, Ziegler-Graff V, Reutenauer A, Herrbach E, Garaud JC, Guilley H, Richards K, Jonard G (1995) Aphid transmission of beet western yellows luteovirus requires the minor capsid read-through protein P74. EMBO J 14:650–659
Brault V, Mutterer J, Scheidecker D, Simonis MT, Herrbach E, Richards K, Ziegler-Graff V (2000) Effects of point mutations in the readthrough domain of the beet western yellows virus minor capsid protein on virus accumulation in planta and on transmission by aphids. J Virol 74:1140–1148
Brault V, Périgon S, Reinbold C, Erdinger M, Scheidecker D, Herrbach E, Richards K, Ziegler-Graff V (2005) The polerovirus minor capsid protein determines vector specificity and intestinal tropism in the aphid. J Virol 79:9685–9693
Bushnell B (2016) BBTools: a suit of bioinformatic tools used for DNA and RNA sequence data analysis. [WWW document] URL http://jgi.doe.gov/data-and-tools/bbtools/.
Chin L-S, Foster JL, Falk BW (1993) The beet western yellows virus ST9-associated RNA shares structural and nucleotide sequence homology with carmo-like viruses. Virology 192:473–482
Congdon B, Matson P, Begum F, Kehoe M, Coutts B (2019) Application of loop-mediated isothermal amplification in an early warning system for epidemics of an externally sourced plant virus. Plants 8:139
Congdon BS, Kehoe MA, Filardo FF, Coutts BA (2019) In-field capable loop-mediated isothermal amplification detection of Turnip yellows virus in plants and its principal aphid vector Myzus persicae. J Virol Methods 265:15–21
Coutts BA, Hawkes JR, Jones RAC (2006) Occurrence of Beet western yellows virus and its aphid vectors in over-summering broad-leafed weeds and volunteer crop plants in the grainbelt region of south-western Australia. Aust J Agric Res 57:975–982
Coutts BA, Jones RAC, Umina P, Davidson J, Baker G, Aftab M (2015) Beet western yellows virus (Synon: Turnip yellows virus) and green peach aphid in canola. South Australian GRDC Updates, Canberra, Australia. https://grdc.com.au/resources-and-publications/grdc-update-papers/tab-content/grdc-update-papers/2015/02/beet-western-yellows-virus-synonym-turnip-yellows-virus-and-green-peach-aphid-in-canola
DeBlasio SL, Johnson R, Mahoney J, Karasev A, Gray SM, MacCoss MJ, Cilia M (2015) insights into the polerovirus-plant interactome revealed by coimmunoprecipitation and mass spectrometry. Mol Plant-Microbe Interact 28:467–481
Dombrovsky A, Glanz E, Lachman O, Sela N, Doron-Faigenboim A, Antignus Y (2013) The complete genomic sequence of pepper yellow leaf curl virus (PYLCV) and its implications for our understanding of evolution dynamics in the genus polerovirus. PLoS ONE 8:e70722–e70722
Domingo E, Baranowski E, Ruiz-Jarabo CM, Martín-Hernández AM, Sáiz JC, Escarmís C (1998) Quasispecies structure and persistence of RNA viruses. Emerg Infect Dis 4:521–527
Duffus JE, Russell GE (1970) Serological and host range evidence for the occurence of beet western yellows virus in Europe. Phytopathology 60:1199–1202
Falk BW, Chin L-S, Duffus JE (1989) Complementary DNA cloning and hybridization analysis of beet western yellows luteovirus RNAs. J Gen Virol 70:1301–1309
Falk BW, Duffus JE (1984) Identification of small single- and double-stranded RNAs associated with severe symptoms in beet western yellows virus-infected capsella bursa-pastoris. Phytopathology 74:1224–1229
Fiallo-Olivé E, Navas-Hermosilla E, Ferro CG, Zerbini FM, Navas-Castillo J (2018) Evidence for a complex of emergent poleroviruses affecting pepper worldwide. Arch Virol 163:1171–1178
Filardo FF, Sharman M (2019) Siratro latent polerovirus (SLPV): a new polerovirus from Australia with a non-functional open reading frame 0. Australas Plant Path 48:491–501
Filardo FF, Thomas JE, Webb M, Sharman M (2019) Faba bean polerovirus 1 (FBPV-1); a new polerovirus infecting legume crops in Australia. Arch Virol 164:1915–1921
Freeman A, Aftab M (2011) Effective management of viruses in pulse crops in south eastern Australia should include management of weeds. Australas Plant Path 40:430–441
Gray S, Cilia M, Ghanim M (2014) Chapter four - circulative, “nonpropagative” virus transmission: an orchestra of virus-, insect-, and plant-derived instruments. In: Maramorosch K, Murphy FA (eds) Advances in virus research. Academic Press, Cambridge, pp 141–199
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Harrison BD (2002) Virus variation in relation to resistance-breaking in plants. Euphytica 124:181–192
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2017) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35:518–522
Huson DH, Bryant D (2005) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jaag HM, Kawchuk L, Rohde W, Fischer R, Emans N, Prüfer D (2003) An unusual internal ribosomal entry site of inverted symmetry directs expression of a potato leafroll polerovirus replication-associated protein. P Natl Acad Sci 100:8939–8944
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8:275–282
Katul L (1992) Characterization by serology and molecular biology of bean leaf roll virus and faba bean necrotic yellows virus. PhD thesis, University of Göttingen, Göttingen, Germany, p 115
King AM, Lefkowitz E, Adams MJ, Carstens EB (2011) Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier, Amsterdam
Kinoti WM, Nancarrow N, Dann A, Rodoni BC, Constable FE (2020) Updating the quarantine status of prunus infecting viruses in Australia. Viruses 12:246
Kruger F (2015) Trim Galore. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
Kuhn JH, Wolf YI, Krupovic M, Zhang Y-Z, Maes P, Dolja VV, Koonin EV (2019) Classify viruses—the gain is worth the pain. Nature 566:318–320
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lim S, Yoo RH, Igori D, Zhao F, Kim KH, Moon JS (2015) Genome sequence of a recombinant brassica yellows virus infecting Chinese cabbage. Arch Virol 160:597–600
Liu M, Liu X, Li X, Zhang D, Dai L, Tang Q (2016) Complete genome sequence of a Chinese isolate of pepper vein yellows virus and evolutionary analysis based on the CP, MP and RdRp coding regions. Arch Virol 161:677–683
Lotos L, Olmos A, Orfanidou C, Efthimiou K, Avgelis A, Katis NI, Maliogka VI (2017) Insights into the etiology of polerovirus-induced pepper yellows disease. Phytopathology 107:1567–1576
MacKenzie DJ, McLean MA, Mukerji S, Green M (1997) Improved RNA extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Dis 81:222–226
Maina S, Edwards OR, Jones RAC (2016) First complete genome sequence of Pepper vein yellows virus from Australia. Genome Announc 4:e00450-e1416
Makkouk KM, Comeau A (1994) Evaluation of various methods for the detection of barley yellow dwarf virus by the tissue-blot immunoassay and its use for virus detection in cereals inoculated at different growth stages. Eur J Plant Pathol 100:71
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003–vev003
Mayo MA, D’Arcy CJ (1999) Family Luteoviridae: a reclassification of Luteoviruses. CABI publishing, Wallingford
Mayo MA, Miller WA (1999) The structure and expression of luteovirus genomes. In: Smith HG, Barker H (eds) The Luteoviridae. CAB International, Wallingford, pp 23–42
Mayo MA (2002) virology division news: ICTV at the Paris ICV: results of the plenary session and the binomial ballot. Arch Virol 147:2254–2260
Minh BQ, Hahn MW, Lanfear R (2020) New methods to calculate concordance factors for phylogenomic datasets. Mol Biol Evol 37:2727–2733
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277–e108277
New S-A, van Heerden SW, Pietersen G, Esterhuizen LL (2016) First report of a turnip yellows virus in association with the brassica stunting disorder in South Africa. Plant Dis 100:2341
Newbert M (2016) The genetic diversity of Turnip yellows virus in oilseed rape (Brassica napus) in Europe, pathogenic determinants, new sources of resistance and host range. Doctoral dissertation, University of Warwick, Coventry. http://webcat.warwick.ac.uk/record=b2869367~S1
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2014) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Pagán I, Holmes EC (2010) Long-term evolution of the luteoviridae: time scale and mode of virus speciation. J Virol 84:6177–6187
Passmore BK, Sanger M, Chin LS, Falk BW, Bruening G (1993) Beet western yellows virus-associated RNA: an independently replicating RNA that stimulates virus accumulation. P Natl Acad Sci 90:10168–10172
Pazhouhandeh M, Dieterle M, Marrocco K, Lechner E, Berry B, Brault V, Pazhouhandeh M, Dieterle M, Marrocco K, Lechner E, Berry B, Brault V, Xe R, Hemmer O, Kretsch T, Richard KE, Genschik P, Ziegler-Graff V, Xe R (2006) F-box-like domain in the polerovirus protein P0 is required for silencing suppressor function. Proc Natl Acad Sci USA 103:1994–1999
Peter KA, Liang D, Palukaitis P, Gray SM (2008) Small deletions in the potato leafroll virus readthrough protein affect particle morphology, aphid transmission, virus movement and accumulation. J Gen Virol 89:2037–2045
Peter KA, Gildow F, Palukaitis P, Gray SM (2009) The C terminus of the polerovirus P5 readthrough domain limits virus infection to the phloem. J Virol 83:5419–5429
Prüfer D, Kawchuk LM, Rohde W (2006) Polerovirus ORF0 genes induce a host-specific response resembling viral infection. Can J Plant Path 28:302–309
Sánchez PAG, Mesa HJ, Montoya MM (2016) Next generation sequence analysis of the forage peanut (Arachis pintoi) virome. Revista Facultad Nacional de Agronomía Medellín 69:7881–7891
Sanger M, Passmore B, Falk BW, Bruening G, Ding B, Lucas WJ (1994) Symptom severity of beet western yellows virus strain ST9 is conferred by the ST9-associated RNA and is not associated with virus release from the phloem. Virology 200:48–55
Schliephake E, Graichen K, Rabenstein F (2000) Investigations on the vector transmission of the Beet mild yellowing virus (BMYV) and the Turnip yellows virus (TuYV). Zeitschrift für Pflanzenkrankheiten und Pflanzenschutz 107:81–87
Schmitz J, Stussi-Garaud C, Tacke E, Prüfer D, Rohde W, Rohfritsch O (1997) In situlocalization of the putative movement protein (pr17) from Potato leafroll luteovirus (PLRV) in infected and transgenic potato plants. Virology 235:311–322
SeedWorld (2019) Turnip yellows virus: the underestimated threat in oilseed rape cultivation. https://seedworld.com/turnip-yellows-virus-the-underestimated-threat-in-oilseed-rape-cultivation/
Shahraeen N, Farzadfar S, Lesemann D-E (2003) Incidence of viruses infecting winter oilseed rape (Brassica napus ssp. oleifera) in Iran. J Phytopathol 151:614–616
Sharman M, Lapbanjob S, Sebunruang P, Belot JL, Galbieri R, Giband M, Suassuna N (2015) First report of Cotton leafroll dwarf virus in Thailand using a species-specific PCR validated with isolates from Brazil. Australas Plant Dis Notes 10:1–4
Shukla DD, Ward CW (1988) Amino acid sequence homology of coat proteins as a basis for identification and classification of the potyvirus group. J Gen Virol 69:2703–2710
Simon-Loriere E, Holmes EC (2011) Why do RNA viruses recombine? Nat Rev Microbiol 9:617–626
Smirnova E, Firth AE, Miller WA, Scheidecker D, Brault V, Reinbold C, Rakotondrafara AM, Chung BYW, Ziegler-Graff V (2015) Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog 11:e1004868
Stevens M, Smith HG, Hallsworth PB (1995) Detection of the luteoviruses, beet mild yellowing virus and beet western yellows virus, in aphids caught in sugar-beet and oilseed rape crops, 1990–1993. Ann Appl Biol 127:309–320
Stevens M, McGrann G, Clark B, Authority H (2008) Turnip yellows virus (syn Beet western yellows virus): An emerging threat to European oilseed rape production. Research Review 69 HGCA. Available via: http://www.hgca.com/document.aspx?fn=load&media_id=4473&publicationId=4579
van den Heuvel JF, Bruyère A, Hogenhout SA, Ziegler-Graff V, Brault V, Verbeek M, van der Wilk F, Richards K (1997) The N-terminal region of the luteovirus readthrough domain determines virus binding to Buchnera GroEL and is essential for virus persistence in the aphid. J Virol 71:7258–7265
van der Wilk F, Verbeek M, Dullemans AM, van den Heuvel JFJM (1997) The genome-linked protein of potato leafroll virus is located downstream of the putative protease domain of the ORF1 product. Virology 234:300–303
van Leur JAG, Aftab M, Manning W, Bowring A, Riley MJ (2013) A severe outbreak of chickpea viruses in northern New South Wales, Australia, during 2012. Australas Plant Dis Notes 8:49–53
Wang F, Wu QF, Zhou BG, Gao ZL, Xu DF (2015) First Report of Turnip yellows virus in Tobacco in China. Plant Dis 99:1870–1870
Worobey M, Holmes EC (1999) Evolutionary aspects of recombination in RNA viruses. J Gen Virol 80:2535–2543
Xiang H-Y, Dong S-W, Shang Q-X, Zhou C-J, Li D-W, Yu J-L, Han C-G (2011) Molecular characterization of two genotypes of a new polerovirus infecting brassicas in China. Arch Virol 156:2251–2255
Yoshida N, Tamada T (2019) Host range and molecular analysis of Beet leaf yellowing virus, Beet western yellows virus-JP and Brassica yellows virus in Japan. Plant Pathol 68:1045–1058
Zhang X-Y, Xiang H-Y, Zhou C-J, Li D-W, Yu J-L, Han C-G (2014) Complete genome sequence analysis identifies a new genotype of brassica yellows virus that infects cabbage and radish in China. Arch Virol 159:2177–2180
Ziegler-Graff V, Brault V, Mutterer JD, Simonis MT, Herrbach E, Guilley H, Richards KE, Jonard G (1996) The coat protein of beet western yellows luteovirus is essential for systemic infection but the viral gene products P29 and P19 are dispensable for systemic infection and aphid transmission. Mol Plant Microbe In 9(6):501–510
Acknowledgements
This study was funded by the Queensland Department of Agriculture and Fisheries and the Australian Grains Research and Development Corporation projects DAN00202, DAQ00186 and DAQ00154. We thank Dr. Joop Van Leur and Dr. Brenda Coutts for discussions about the project and assistance with sample collection, and Dr. Paul Campbell for discussions about the results.
GenBank accession numbers MT586571– MT586598 and MT642436–MT642445.
Funding
This study was funded by the Queensland Department of Agriculture and Fisheries and the Australian Grains Research and Development Corporation (projects DAN00202, DAQ00186 and DAQ00154).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there are no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Jesús Navas-Castillo.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Filardo, F., Nancarrow, N., Kehoe, M. et al. Genetic diversity and recombination between turnip yellows virus strains in Australia. Arch Virol 166, 813–829 (2021). https://doi.org/10.1007/s00705-020-04931-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04931-w