
Abstract
We investigated and analysed the molecular evolution of hemagglutinin (HA) and neuraminidase (NA) of influenza A(H1N1)pdm09 virus during the 2017–2018 and 2018–2019 influenza seasons in Beijing, China. We collected and extracted RNA from influenza A(H1N1)pdm09 strains from Peking University People’s Hospital and analyzed their HA and NA genes by RT-PCR and sequencing. Phylogenetic analysis of HA and NA sequences was used to compare the amino acid sequences of 51 strains with those of reference strains. All strains belonged to subclade 6B.1, with S162N and I216T substitutions (H1 numbering). Our strains differed from strain A/Michigan/45/2015, with the substitutions S91R, S181T and I312V in the HA antigenic epitope. An E189G mutation was detected in the 190 helix of the receptor binding region of HA. A new potential glycosylation site, 179 (NQT), which was not detected before the 2015 influenza season, was identified. Two strains were mutated at I223, the NA inhibitor resistance site. During 2012-2019, amino acids of HA and NA mutated over time. Co-occurrence mutations N146D, S200P, S202I and A273T in HA appeared along with Q51K, F74S and D416N in NA in six strains during two influenza seasons. Our work reveals the molecular changes and phylogenetic characteristics of influenza A(H1N1)pdm09 virus and suggests that a vaccine probably provides suboptimal protection. The biological characteristics of the new glycosylation and drug-resistance sites detected in this work need to be studied further. The co-occurrence of mutations in HA and NA might affect the characteristics of the virus and need to be given more attention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Influenza is a common respiratory infection caused by influenza virus. Influenza has been a serious threat to public health worldwide since its emergence. According to the World Health Organization (WHO), the annual epidemic of seasonal human influenza virus can affect 5-10% of adults, causing approximately 3 to 5 million cases of serious disease and approximately 290,000 to 650,000 deaths (WHO, 2018). Influenza is ranked first among the world's top 10 public health threats [27]. Since 2009, when the influenza H1N1 virus caused the first influenza pandemic of the 21st century, the influenza A(H1N1)pdm09 virus has become one of the main subtypes of seasonal influenza viruses. Hemagglutinin (HA) and neuraminidase (NA) are two important proteins on the surface of influenza virus. Since the gene sequence of HA is prone to mutation, HA gene mutations are one of the primary causes of variation in the antigenicity and pathogenicity of the virus [14]. NA plays a key role in the replication and transmission of influenza virus, and NA gene mutations are closely associated with resistance to antiviral drugs [15].
Vaccination is the most effective measure to prevent influenza, and neuraminidase inhibitors (NAIs) are effective drugs to treat influenza. In 2017, WHO recommended using the A/Michigan/45/2015 strain to replace the A/California/07/2009 strain in influenza A(H1N1)pdm09 vaccines for the Northern Hemisphere, suggesting a certain antigenic drift of influenza A(H1N1)pdm09 virus compared with the previous period [1, 2]. Although WHO updates the universal influenza vaccine every few years, a mismatch between influenza virus vaccine strains and epidemic strains still exists. The influenza vaccine can effectively protect only 50%-60% of the population, and the coverage rate of the influenza vaccine in mainland China is only 23.2% [31]. In addition, some mutant influenza H1N1 viruses are resistant to adamantanes and NAIs [12, 30]. These data indicate that analysing the dynamics of molecular evolution and antigenic mutations is important for predicting influenza epidemics and assessing the effectiveness of vaccines and drugs.
In the present study, we collected and extracted RNA of influenza A(H1N1)pdm09 strains isolated from Peking University People’s Hospital (PKUPH) during the influenza epidemic seasons from November 2017 to March 2018 and from November 2018 to March 2019. We mainly examined the molecular evolution and genetic properties of the HA and NA genes of isolates from in Beijing, China, which has not been reported previously. The findings of our study will help us understand the evolution process and mutations of HA and NA genes of the influenza H1N1 virus more comprehensively and provide a basis for the prediction and prevention of influenza epidemics.
Materials and methods
Case definition and nasal specimens
The study population included patients with influenza-like illness (ILI) admitted to PKUPH from November 2017 to March 2018 and from November 2018 to March 2019. ILI is defined in strict accordance with WHO standards as an acute respiratory illness with a measured temperature of ≥ 38 °C and cough, with onset within the preceding 10 days [11]. Nasal swabs were immediately placed in virus transport medium tubes and stored at -80 °C until analysis. A total of 32,240 ILI nasal swabs were collected.
Data collection
The Chinese National Influenza Center (CNIC) monitors patients with ILI nationwide through its Influenza Laboratory Surveillance Network, which collects and tests thousands of specimens from ILI patients in sentinel hospitals nationwide on a weekly basis. We summarized the weekly CNIC data of ILI and confirmed influenza cases during the influenza epidemic from July 2017 to June 2019 to understand the epidemic situation of influenza in North China. We also collected HA and NA gene sequences of influenza A(H1N1)pdm09 strains isolated from PKUPH during the period from 2012 to 2017.
Viral RNA extraction, gene amplification, and sequencing
We extracted viral RNA from 140 μl of nasal swabs, using a QIAamp Viral RNA Mini Kit (QIAGEN, Germany) according to the manufacturer’s instructions. Reverse transcription was performed at 50 °C for 50 min using the universal primer 5’-AGCAAAAGCAGGTAG-3’ [35]. For detection of influenza A and B viruses, we used the forward primer and the reverse primer for amplification of the matrix gene [9] (Table 1). Amplification reactions were carried out under the following conditions: 95 °C for 15 min; followed by 40 cycles of 94 °C for 30 s, 46 °C for 15 s, and 72 °C for 30 s; and then 42 °C for 10 s.
We used H1- and N1-specific primers to amplify HA and NA genes of the influenza A(H1N1)pdm09 viruses [6] (Table 1). The cycling conditions were 94 °C for 3 min; followed by 40 cycles of 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 1.5 min; and a final extension step at 72 °C for 7 min. Fifty-one A(H1N1)pdm09 virus strains were detected, and their products were sequenced using the Sanger method. We obtained a total of 38 HA and 46 NA sequences, and all sequences were deposited in the NCBI GenBank database (GenBank accession numbers: MN853421-MN853458 and MN853485-MN853530).
Phylogenetic analysis
For analysis of the molecular evolution of influenza A(H1N1)pdm09 virus, we determined the HA and NA gene sequences of strains from PKUPH as described above and compared them with other strains collected in previous years. The reference sequences were obtained from NCBI. For each year, from the large collection of global isolates, 2-3 sequences were randomly selected during 2009–2019. A total of 38 HA and 46 NA sequences from PKUPH (Beijing) isolates and 24 HA and 24 NA sequences from global strains were sampled independently for our phylogenetic analysis. ClustalW 2.1 was used for multiple sequence alignments. Phylogenetic analysis of gene segments was performed using MEGA 7.0 software and Tamura’s 3-parameter model with gamma-distributed rates. The reliability of the maximum-likelihood tree was assessed by bootstrap analysis with 1000 replications [18, 29].
Characterization of HA and NA sequences
The A/California/7/2009 and A/Michigan/45/2015 strains were used as the reference strains in this study. All 38 HA and 46 NA amino acid sequences from the PKUPH (Beijing) strains were aligned with their homologs from various countries using MEGA 7.0. All amino acid mutation sites were confirmed, and key mutation sites were screened.
Results
Distribution of influenza
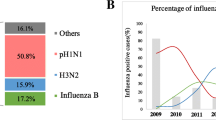
In this study, we collected data from 32,240 ILI patients treated by the Department of Infectious Disease of PKUPH from November 2017 to March 2019. By RT-PCR, we determined that 2978 ILI patients were positive for influenza virus, 2441 (82.0%) of whom were positive for influenza A virus, while 537 (18.0%) were positive for influenza B virus. We collected data for ILI and confirmed influenza cases in northern China from July 2017 to June 2019 to determine the distribution of influenza. The distribution of ILI cases was consistent with that of confirmed influenza cases. According to the CNIC, during the 2017-2018 influenza season in Beijing, influenza A(H1N1)pdm09 and A(H3N2) viruses co-circulated with the predominance of influenza B virus, with a peak period that commenced at the 47th week of 2017 and lasted approximately 2 months (Fig. 1). However, during the 2018-2019 influenza season in Beijing, influenza A(H1N1)pdm09 virus became the completely dominant subtype in the first peak period, and subsequently, the number of cases of influenza B increased rapidly; influenza A(H1N1)pdm09, A(H3N2), and B viruses constituted the second peak (Fig. 1).

The epidemic situation in northern China from July 2017 to June 2019. Influenza B virus was the most dominant subtype during the 2017-2018 influenza season, while the influenza A(H1N1)pdm09 virus became the completely predominant subtypes in the peak phase of the 2018-2019 influenza season. The influenza data from northern China was obtained from the Chinese National Influenza Center.
Sequence comparisons
Among the Beijing influenza A(H1N1)pdm09 strains from PKUPH obtained during the 2017–2018 and 2018-2019 influenza seasons, we found that the nucleotide sequence identity values for of HA and NA were 96.4%-99.4% and 92.2%-100%, respectively, and the amino acid sequence identity values for HA and NA were 96.1%-99.3% and 91.3%-99.6%, respectively. When comparing the gene sequences of the 51 strains of influenza A(H1N1)pdm09 virus from this study with those of the influenza A/California/07/2009 reference strain, we observed that the HA nucleotide and amino acid sequences were 94.7%-96.6% and 94.2%-96.3% identical, respectively; and the NA nucleotide and amino acid sequences were 90.3%-97.1% and 88.1%-95.3% identical, respectively. When comparing the gene sequences of the 51 strains of influenza A(H1N1)pdm09 virus from this study with those of the influenza A/Michigan/45/2015 vaccine strain, the HA nucleotide and amino acid sequences were 96.5%-98.6% and 96.6%-98.9% identical, respectively, and the NA nucleotide and amino acid sequences were 92.1%-99.0% and 90.9%-98.9% identical, respectively. The isolates obtained from PKUPH were more similar to the influenza A/Michigan/45/2015 strain than to the influenza A/California/07/2009 strain.
Evolutionary and mutational analysis of influenza A(H1N1)pdm09 virus
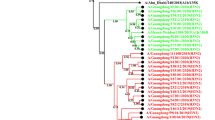
We constructed phylogenetic trees for the HA and NA genes of the 51 strains of influenza A(H1N1)pdm09 virus from PKUPH (Beijing), the vaccine strains A/California/07/2009 and A/Michigan/45/2015, and the viruses circulating in other regions of the world (Figs. 2 and 3). HA phylogenetic analysis showed that all PKUPH (Beijing) strains formed a distinct cluster, belonging to subclade 6B.1 (Fig. 2). This subclade includes the currently recommended vaccine virus A/Michigan/45/2015 with the amino acid substitutions S162N and I216T (potential gain of glycosylation; H1 numbering) [7]. However, all of the isolates differed from the vaccine virus A/Michigan/45/2015, due to the HA antigenic epitope substitutions S91R, S181T and I312V (H1 numbering), indicating that the isolates belong to the 6B.1A subclade. Additionally, differences were found between strains isolated from November 2017 to March 2018 and from November 2018 to March 2019. The former group was defined by the amino acid substitutions Y10C, D52N, H155Y and A273I (H1 numbering).

Phylogenetic tree based on HA nucleotide sequences of influenza A(H1N1)pdm09 virus from Beijing and global strains reported from 2009 to 2019. “▲” represents the vaccine strains. “●” represents the isolates from this study. The tree was constructed by the maximum-likelihood method in MEGA 7.0 with 1000 bootstrap replicates; nodes with bootstrap values >70 are shown. The 2017–2019 Beijing strains are indicated in different colours (strains from 2017 in the 2017-2018 influenza season are shown in red, strains from 2018 in the 2017-2018 influenza season are shown in yellow, strains from 2018 in the 2018-2019 influenza season are shown in blue, and strains from 2019 in the 2018-2019 influenza season are shown in green).

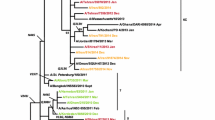
Phylogenetic tree based on NA nucleotide sequences of influenza A(H1N1)pdm09 virus from Beijing and global strains reported from 2009 to 2019. “▲” represents the vaccine strain. “●” represents the isolates from this study. The tree ws constructed by the maximum-likelihood method in MEGA 7.0 with 1000 bootstrap replicates; nodes with bootstrap values >70 are shown. The 2017–2019 Beijing strains are indicated in different colours (strains from 2017 in the 2017-2018 influenza season are shown in red, strains from 2018 in the influenza 2017-2018 season are shown in yellow, strains from 2018 in the 2018-2019 influenza season are shown in blue, and strains from 2019 in the 2018-2019 influenza season are shown in green).
A similar evolutionary characteristic was observed when analysing the NA sequences (Fig. 3). The NA nucleotide sequences of the 2018–2019 viruses displayed a drift relative to the 2017–2018 strain. All of the isolates were similar to epidemic strains from the USA, Germany, France and South Korea that were isolated during the same period.
Analysis of amino acid sequence variations in the HA and NA proteins
In this study, we compared the amino acid sequences of the HA protein between the PKUPH (Beijing) A(H1N1)pdm09 strains and vaccine strain A/Michigan/45/2015. The entire article follows the H1 numbering system. All isolates had the S91R, S181T and I312V mutations, which were the characteristic mutations of the influenza A(H1N1)pdm09 virus of these two seasons (Table 2). In the antigenic epitopes, we found that all isolates of the 2017‐2018 and 2018-2019 seasons harboured the following mutations: S181T (100%) in the Sa site; T202I (28.2%), a less common mutation, in the Sb site; H155Y (23.1%), a less common mutation, in the Ca site; and S91R (100%), a common mutation, in the Cb site (Table 3). In addition, L178I (2.5%) and P154S (2.5%), rare mutations that were detected in one isolate, were found in the Sa site and Cb site, respectively. The receptor-binding region (RBD) of HA consists of three domains, including the 130 loop, the 220 loop, and the 190 helix [23], which are associated with adaptability to α2–6-linked glycans, which are receptors for human influenza viruses In the RBD of HA, 3.9% of isolates had the E189G mutation, and other sites did not mutate. D222G is an additional mutation in HA that has been reported to enhance the binding to α2–6-linked glycans in the human lower respiratory tract [8]. However, we did not observe this mutation in the isolated A(H1N1)pdm09 strains. We detected nine potential glycosylation sites in the HA gene: NNS27-29, NST28-30, NVT40-42, NGT104-106, NQT179-181, NXT293-295, NST304-306, NGT498-500, and NGS557-559. At the potential glycosylation sites, NTT at amino acid 293 was mutated to NAT in a small number of strains (5.1%). We also found that all of the strains had a new glycosylation site 179 (NQT), which is located in the antigenic epitope. The 179 glycosylation site began to appear in 2015 and became the main feature of all strains isolated in the following years.
We analysed the NA protein sequences of the PKUPH (Beijing) A(H1N1)pdm09 strains and compared them to the NA protein sequence of the vaccine strain A/Michigan/45/2015. The alignment of the NA amino acid sequences showed that all isolated strains had the amino acid substitutions V81A and N449D (Table 2). We detected twelve mutations that were in the NA antigenic sites, which are represented by NA residues 83–143 (I99V, I117M), 156–190 (I188T, N189S), 252–303 (V267A, G298V), 330, 332, 340–345, 368, 370, 387–395 (I389K), 400 (S400T), 431–435, and 448–468 (N449D, S450R, D451N, D451E) [4] (Table 4). The catalytic sites in the NA protein include R118, D151, R152, R225, E277, R293, R368, and Y402, and the framework sites supporting the catalytic residues include E119, R156, W179, S180, D/N199, I223, E228, H275, E278, N295 and E425 [4, 26]. However, most of these residues were highly conserved in strains isolated from the 2017–2018 and 2018-2019 influenza seasons. The binding site of the NA neutralizing antibody is the structural epitope around the catalytic centre of NA. Around that location, we found that one strain had a rare I177M mutation, and all of the strains had the common N369K mutation. In addition, we observed that three potential N-linked glycosylation sites had variations in NA (Table 5). Importantly, two strains had an I223 mutation, which is associated with reduced susceptibility to several NAIs [24]. None of the analysed strains had the oseltamivir resistance mutation H275Y.
Annual changes of amino acid substitutions
We collected HA and NA gene sequences of influenza A(H1N1)pdm09 strains from the data bank of PKUPH during the period from 2012 to 2017 and compared the gene sequences from 2012-2017 and 2017-2019 to the vaccine strains A/California/07/2009 and A/Michigan/45/2015. In our study, for HA, the amino acid substitution H155Y appeared in the 2017-2018 influenza season and disappeared in the 2018-2019 influenza season, representing a transient change (Table 6). At site 202, Thr mutated to Ile, which was detected in the 2017-2018 and 2018-2019 influenza seasons (Table 6). The I233T mutation has been extant since 2017 and has been a feature of subsequent seasons (Table 6). The changes of other amino acid substitutions in the HA protein sequence are shown in Table 6, including sites 146, 200, 273 and 421.
From 2012 to 2019, changes in amino acid substitutions in the NA protein are shown in Table 7. Among them, mutations at sites 51, 74, 77, 188, 314 and 416 appeared in the 2017-2018 influenza season. Mutations at sites 189 and 389 followed, appearing since the 2018-2019 influenza season (Table 7). During these seven seasons, amino acids at sites 45, 449, 450 and 451 have changed constantly (Table 7).
The N146D, S200P, S202I and A273T mutations in HA appeared along with Q51K, F74S and D416N mutations in NA in six strains during the 2017–2018 and 2018-2019 influenza seasons. H155Y and A273I in HA were detected in nine strains in the same period (Tables 5 and 6).
Discussion
Since the influenza pandemic of 2009, the influenza A(H1N1)pdm09 virus has gradually replaced the seasonal influenza A(H1N1) virus and become one of the main subtypes of the influenza epidemic in winter and spring worldwide. Influenza A(H1N1)pdm09 virus adapts to new environments and hosts by constantly changing [13]. Each country has a different start time and major subtype of the influenza season each year. For example, the distribution of influenza strain subtypes in the Northern Hemisphere during the 2017–2018 influenza season indicates that the influenza epidemiology of China was similar to that of other regions in Asia but was different from that of North America and European countries [2].
The influenza vaccine is the primary measure to prevent influenza virus infections. Each year, WHO collects data on the antigenic characteristics of circulating viruses and analyzes the evolutionary history of HA to choose influenza vaccine strains for the Northern and Southern Hemispheres. Sequence similarity comparisons revealed that the isolates from 2017 to 2019 in this study were divergent from the vaccine strain A/Michigan/45/2015 and that the similarity between the isolates and the vaccine virus decreased year by year. All of the isolates differed from the vaccine virus A/Michigan/45/2015, with the HA antigenic epitope substitutions S91R, S181T and I312V, potentially indicating that the surface antigen of these strains had constantly mutated and gradually evolved. This suggests that the current vaccines probably provide only moderate protection against the influenza A(H1N1)pdm09 virus, and such protection might have been reduced or even absent in the last season.
During infection, HA binds to sialic acid receptors on the host cell surface and allows the virus to enter the cell by mediating membrane fusion [32]. We found some common and less common mutations in HA, which may lead to changes in the antigen specificity, host adaptability, and pathogenicity of influenza virus. The HA protein of the influenza A(H1N1)pdm09 virus has four antigenic epitopes, Sa, Sb, Ca and Cb [21]. Amino acid substitutions were detected in these four antigenic sites. Studies have shown that neutralizing antibodies protect against influenza virus infection and transmission by blocking the binding of HA to cell receptors and inhibiting membrane fusion [33]. Therefore, amino acid substitutions in of HA antigen epitopes may result in immune escape by the virus. In addition, other mutations (V169I, E189G and S200P) are close to antigenic sites and therefore may indirectly influence antigenicity and virulence. Interestingly, we found that the fixed mutation S202T was replaced by S202I in eleven (28%) isolated strains. Whether this change (Thr to Ile) alters the fitness of influenza virus remains to be investigated.
The NA protein catalyses the hydrolysis of sialic acid, which reduces the affinity of new virus particles for the host cell and promotes the release of the virus [20]. Currently, NAIs are the most effective treatment for influenza. The catalytic sites and framework sites of NA are usually conserved, but mutations at these sites, such as H275Y, I223R and E119G, directly affect the effect of NAIs [15]. In our study, we detected a drug-resistance mutation, I223R, in one strain that was isolated from a patient with nephrotic syndrome who had been taking an immunosuppressor for a long time. The mechanism of resistance to oseltamivir was due to the loss of key hydrogen bonds between the inhibitor and residues in the 150-loop [34]. When the I223R mutation in NA occurs, the structure of the NA protein transitions from a closed conformation to an open conformation. Oseltamivir binds weakly to the open conformation of NA. Although the influenza A(H1N1)pdm09 virus with H275Y in NA is defective in its biological characteristics, drug-resistance mutations (E119D, H275Y and I223R) are often detected in viruses from immunosuppressed populations [19, 28, 30]. Therefore, these mutated viruses may need a friendly environment to survive. This might explain why influenza viruses are more susceptible to drug-resistance mutations in immunosuppressed populations.
We also found some mutations that change over time by comparing the relevant data of PKUPH over the last seven years. Glycosylation of the HA head antigen sites not only changes the properties of the HA protein but also increases the pathogenicity of the virus by hiding it from immune recognition [5]. We detected a new potential glycosylation site, NQT, at site 179, which has been reported in studies from other countries [17]. The glycosylation of site 179 did not appear in China before 2015 [9, 10] but became the main feature of all strains isolated during the 2015-2019 influenza seasons. The appearance of this glycosylation site may be related to vaccine pressure. Interestingly, it has been reported that no amino acid substitutions occurred at I233 of the HA protein of the influenza A(H1N1)pdm09 virus in China before 2017; however, in our study, the I233T mutation in HA has been extant since 2017 and has been a feature in subsequent seasons (Table 6). It is known that the I233 mutation can change the structure of the HA protein and reduce its pH stability. By disrupting hydrophobic interactions between I233 and V237, mutations at position 233 may reduce the pH stability of the HA protein. Such intramolecular changes may alter the conformation of a loop formed by amino acids 230–242, thereby reducing receptor specificity [16]. Whether the I233T mutation affects the adaptability of the virus requires further study.
Furthermore, co-occurrence of mutations in the HA and NA genes in the influenza A(H1N1)pdm09 virus were detected in this study, suggesting that these substitutions occurred simultaneously. The amino acids substitutions S220T in HA and N248D in NA were found in our study, and these are the main molecular markers that distinguish the beginning and the peak of the 2009-2010 influenza A(H1N1) season [22]. The N146D, S200P, S202I, A273T and N277D mutations in HA appeared along with the Q51K, F74S and D416N mutations in NA in six strains during the 2017–2019 influenza seasons. Similarly, the co-mutations H155Y and A273I in HA were detected in nine strains in the same period, indicating that co-mutations in HA and NA may not occur independently but are probably associated with each other [8]. Importantly, the newly emerged substitutions (HA: 91, 181 and 312; NA: 77, 81, 188 and 449) in almost all isolates from the 2017-2018 and 2018-2019 influenza seasons were observed in the strains circulating globally from 2017–2019 but were not found in the strains before 2017. Previous studies showed that the viability of the strain with the H275Y mutation in NA decreased to a certain extent, but this strain has persisted in recent years and remained adaptable, because of the compensating effects of other mutations [3, 25]. Co-mutation strains (HA: S91R, S181T and I312V; NA: G77R, V81A, I188T and N449D) may increase the fitness of the virus in a new environment and host. The increased fitness and decreased sequence similarity of co-mutation strains to the A/Michigan/45/2015 strain may lead to a reduced efficacy of the WHO-recommended vaccine. The co-mutations of the HA and NA proteins of the influenza A(H1N1)pdm09 virus need to be studied further, and attention should be paid particularly to the accumulation of mutations that may cause an epidemic of new influenza A(H1N1)pdm09 lineages.
Conclusions
The present work further enhances our understanding of the winter influenza epidemic in China and the Northern Hemisphere and thus provides insights for prediction of the next epidemic strain of the influenza A(H1N1)pdm09 virus. Mutations of HA and NA, including new glycosylation and drug-resistance mutations, were detected in this study, and the biological characteristics of these mutations require further study. Furthermore, co-mutations in the HA and NA proteins, which may affect the adaptability of the influenza virus to the environment and the host, need to be taken seriously, and more research is needed to overcome the influenza problem.
References
World Health Organization (2016) Recommended composition of influenza virus vaccines for use in the 2016–2017 northern hemisphere influenza season. Wkly Epidemiol Rec 91:121–132
World Health Organization (2018) Recommended composition of influenza virus vaccines for use in the 2018–2019 northern hemisphere influenza season. Wkly Epidemiol Rec 93:133–141
Abed Y, Pizzorno A, Bouhy X, Rheaume C, Boivin G (2014) Impact of potential permissive neuraminidase mutations on viral fitness of the H275Y oseltamivir-resistant influenza A(H1N1)pdm09 virus in vitro, in mice and in ferrets. J Virol 88:1652–1658
Air GM (2012) Influenza neuraminidase. Influ Other Respir Viruses 6:245–256
Al Khatib HA, Al Thani AA, Gallouzi I, Yassine HM (2019) Epidemiological and genetic characterization of pH1N1 and H3N2 influenza viruses circulated in MENA region during 2009–2017. BMC Infect Dis 19:314
Barbezange C, Jones L, Blanc H, Isakov O, Celniker G, Enouf V, Shomron N, Vignuzzi M, van der Werf S (2018) Seasonal genetic drift of human influenza A virus quasispecies revealed by deep sequencing. Front Microbiol 9:2596
Chambers C, Skowronski DM, Sabaiduc S, Winter AL, Dickinson JA, De Serres G, Gubbay JB, Drews SJ, Martineau C, Eshaghi A, Krajden M, Bastien N, Li Y (2016) Interim estimates of 2015/16 vaccine effectiveness against influenza A(H1N1)pdm09, Canada, February 2016. Euro Surveill 21:30168
Chan PK, Lee N, Joynt GM, Choi KW, Cheung JL, Yeung AC, Lam P, Wong R, Leung BW, So HY, Lam WY, Hui DC (2011) Clinical and virological course of infection with haemagglutinin D222G mutant strain of 2009 pandemic influenza A (H1N1) virus. J Clin Virol 50:320–324
Fang Q, Gao Y, Chen M, Guo X, Yang X, Yang X, Wei L (2014) Molecular epidemiology and evolution of A(H1N1)pdm09 and H3N2 virus during winter 2012–2013 in Beijing, China. Infect Genet Evol 26:228–240
Fang Q, Gao Y, Chen M, Guo X, Yang X, Wei L (2015) Molecular epidemiology and evolution of influenza A and B viruses during winter 2013–2014 in Beijing, China. J Arch Virol 160:1083–1095
Fitzner J, Qasmieh S, Mounts AW, Alexander B, Besselaar T, Briand S, Brown C, Clark S, Dueger E, Gross D, Hauge S, Hirve S, Jorgensen P, Katz MA, Mafi A, Malik M, McCarron M, Meerhoff T, Mori Y, Mott J, Olivera M, Ortiz JR, Palekar R, Rebelo-de-Andrade H, Soetens L, Yahaya AA, Zhang W, Vandemaele K (2018) Revision of clinical case definitions: influenza-like illness and severe acute respiratory infection. Bull World Health Organ 96:122–128
Garcia V, Aris-Brosou S (2014) Comparative dynamics and distribution of influenza drug resistance acquisition to protein m2 and neuraminidase inhibitors. Mol Biol Evol 31:355–363
Goka EA, Vallely PJ, Mutton KJ, Klapper PE (2014) Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: a systematic review and meta-analysis of epidemiological evidence. Arch Virol 159:3167–3183
Guarnaccia T, Carolan LA, Maurer-Stroh S, Lee RT, Job E, Reading PC, Petrie S, McCaw JM, McVernon J, Hurt AC, Kelso A, Mosse J, Barr IG, Laurie KL (2013) Antigenic drift of the pandemic 2009 A(H1N1) influenza virus in A ferret model. PLoS Pathog 9:e1003354
Hussain M, Galvin HD, Haw TY, Nutsford AN, Husain M (2017) Drug resistance in influenza A virus: the epidemiology and management. Infect Drug Resist 10:121–134
Jones S, Nelson-Sathi S, Wang Y, Prasad R, Rayen S, Nandel V, Hu Y, Zhang W, Nair R, Dharmaseelan S, Chirundodh DV, Kumar R, Pillai RM (2019) Evolutionary, genetic, structural characterization and its functional implications for the influenza A (H1N1) infection outbreak in India from 2009 to 2017. Sci Rep 9:14690
Khandaker I, Suzuki A, Kamigaki T, Tohma K, Odagiri T, Okada T, Ohno A, Otani K, Sawayama R, Kawamura K, Okamoto M, Oshitani H (2013) Molecular evolution of the hemagglutinin and neuraminidase genes of pandemic (H1N1) 2009 influenza viruses in Sendai, Japan, during 2009–2011. Virus Genes 21(11):30168
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
L’Huillier AG, Abed Y, Petty TJ, Cordey S, Thomas Y, Bouhy X, Schibler M, Simon A, Chalandon Y, van Delden C, Zdobnov E, Boquete-Suter P, Boivin G, Kaiser L (2015) E119D neuraminidase mutation conferring pan-resistance to neuraminidase inhibitors in an A(H1N1)pdm09 isolate from a stem-cell transplant recipient. J Infect Dis 212:1726–1734
Medina RA, Garcia-Sastre A (2011) Influenza A viruses: new research developments. Nat Rev Microbiol 9:590–603
Mesman AW, Westerhuis BM, Ten Hulscher HI, Jacobi RH, de Bruin E, van Beek J, Buisman AM, Koopmans MP, van Binnendijk RS (2016) Influenza virus A(H1N1)2009 antibody-dependent cellular cytotoxicity in young children prior to the H1N1 pandemic. J Gen Virol 97:2157–2165
Morlighem JE, Aoki S, Kishima M, Hanami M, Ogawa C, Jalloh A, Takahashi Y, Kawai Y, Saga S, Hayashi E, Ban T, Izumi S, Wada A, Mano M, Fukunaga M, Kijima Y, Shiomi M, Inoue K, Hata T, Koretsune Y, Kudo K, Himeno Y, Hirai A, Takahashi K, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Kawaoka Y, Hayashizaki Y, Ishikawa T (2011) Mutation analysis of 2009 pandemic influenza A(H1N1) viruses collected in Japan during the peak phase of the pandemic. PLoS One 6:e18956
Neumann G, Noda T, Kawaoka Y (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939
Nguyen HT, Fry AM, Gubareva LV (2012) Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther 17:159–173
Pizzorno A, Abed Y, Bouhy X, Beaulieu E, Mallett C, Russell R, Boivin G (2012) Impact of mutations at residue I223 of the neuraminidase protein on the resistance profile, replication level, and virulence of the 2009 pandemic influenza virus. Antimicrob Agents Chemother 56:1208–1214
Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ (2006) The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443:45–49
Shao W, Li X, Goraya MU, Wang S, Chen JL (2017) Evolution of influenza A virus by mutation and re-assortment. Int J Mol Sci 18(8):1650
Tamura D, DeBiasi RL, Okomo-Adhiambo M, Mishin VP, Campbell AP, Loechelt B, Wiedermann BL, Fry AM, Gubareva LV (2015) Emergence of multidrug-resistant influenza A(H1N1)pdm09 virus variants in an immunocompromised child treated with oseltamivir and zanamivir. J Infect Dis 212:1209–1213
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687
Trebbien R, Pedersen SS, Vorborg K, Franck KT, Fischer TK (2017) Development of oseltamivir and zanamivir resistance in influenza A(H1N1)pdm09 virus, Denmark, 2014. Euro Surveill 22(3):30445
Wang Q, Yue N, Zheng M, Wang D, Duan C, Yu X, Zhang X, Bao C, Jin H (2018) Influenza vaccination coverage of population and the factors influencing influenza vaccination in mainland China: a meta-analysis. Vaccine 36:7262–7269
Webster RG, Rott R (1987) Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell 50:665–666
Williams JA, Gui L, Hom N, Mileant A, Lee KK (2018) Dissection of epitope-specific mechanisms of neutralization of influenza virus by intact IgG and Fab fragments. J Virol 92(6):e02006-17
Woods CJ, Malaisree M, Pattarapongdilok N, Sompornpisut P, Hannongbua S, Mulholland AJ (2012) Long time scale GPU dynamics reveal the mechanism of drug resistance of the dual mutant I223R/H275Y neuraminidase from H1N1-2009 influenza virus. Biochemistry 51:4364–4375
Zhou B, Deng YM, Barnes JR, Sessions OM, Chou TW, Wilson M, Stark TJ, Volk M, Spirason N, Halpin RA, Kamaraj US, Ding T, Stockwell TB, Salvatore M, Ghedin E, Barr IG, Wentworth DE (2017) Multiplex reverse transcription-PCR for simultaneous surveillance of influenza A and B viruses. J Clin Microbiol 55:3492–3501
Acknowledgements
We thank all the doctors in the Department of Infectious Disease of PKUPH for recruiting patients and collecting nasal specimens. We also thank the laboratory staff in Peking University Hepatology Institute of PKUPH for their guidance.
Funding
This study was supported by the National Natural Science Foundation of China (no. 81541139). The funding sources were not involved.
Author information
Authors and Affiliations
Contributions
Conceptualization, methodology, and supervision were performed by Yan Gao. Material preparation, data collection, and analysis were performed by Baiyi Liu. Baiyi Liu, Yue Wang, Yafen Liu, Yisi Liu, and Yuanyuan Chen collected nasal specimens for this research. Xu Cong and Ying Ji provided help with experimental techniques. The original draft of the manuscript was written by Baiyi Liu, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethics approval
Informed consent was obtained from all patients before collecting nasal samples. This study followed the Declaration of Helsinki Principles, and ethical approval for the study was obtained from the Research Ethics Committee at PKUPH (IRB No. 2016PHB100-01). All analysed data were anonymized.
Additional information
Handling Editor: Ayato Takada.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, B., Wang, Y., Liu, Y. et al. Molecular evolution and characterization of hemagglutinin and neuraminidase of influenza A(H1N1)pdm09 viruses isolated in Beijing, China, during the 2017–2018 and 2018–2019 influenza seasons. Arch Virol 166, 179–189 (2021). https://doi.org/10.1007/s00705-020-04869-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04869-z