
Abstract
An epizootic hemorrhagic disease virus (EHDV) strain designated YN09-04 was isolated from sentinel cattle in China. The length of its complete genome was 19,344 bp in total, consisting of 10 segments ranging in size from 810 bp (S10) to 3942 bp (S1). Based on phylogenetic analysis of the S2 sequence, YN09-04 clusters with EHDV serotype 7 (EHDV-7) strains form a distinct, well-supported subgroup, indicating that YN09-04 belongs to EHDV-7. However, the origin of the YN09-04 genome is very complex. The S2 and S6 of YN09-04 cluster with those of Japanese EHDV-7 strains, whereas the S1, S3, S4, S5 and S7 of YN09-04 share high nucleotide sequence identity and a close relationship with those of Japanese Ibaraki viruses, and the S8, S9 and S10 nucleotide sequences of YN09-04 are more similar to those of some Australian EHDV strains than to those of other isolates. These results suggest that the genome of YN09-04 likely originated from a reassortment event between EHDV strains that were similar to the current Japanese and Australian strains and that YN09-04 and some EHDVs from Japan and Australia share the same ancestors. This is the first report of the isolation, identification and complete-genome phylogenetic analysis of an EHDV-7 strain from China.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epizootic hemorrhagic disease virus (EHDV) belongs to the genus Orbivirus, family Reoviridae. EHDV contains a double-stranded RNA genome that consists of 10 segments (S1 to S10) and encodes a total of seven structural proteins (VP1-VP7) and four nonstructural proteins (NS1-NS4) [9]. Based on antigenic and genetic analysis of the outer capsid protein VP2, EHDV isolates have been assigned to seven serotypes [3]. Ibaraki virus (IBAV), which belongs to EHDV-2, caused 43,793 infections and 4,298 deaths in cattle in Japan during the period of 1959-1960, in addition, more than 1,000 cases of abortions and stillbirths were observed among IBAV-infected pregnant cows in Japan from 1997 to 1998 [6, 7]. In Israel, EHDV-7 caused a widespread outbreak in cattle, resulting in significant economic losses with 5-80% morbidity, 1% mortality, a 10-20% decline in milk production, and the involuntary culling of a large number of cattle [4, 13]. Orbiviruses have the potential to undergo reassortment between different serotypes of the same species during coinfection under natural and experimental conditions [10, 11]. KSB-14/E/97, which was initially identified as a new IBAV strain and was isolated from cattle with the clinical signs of Ibaraki disease during an IBAV epidemic in Japan, has been reclassified as EHDV-7 based on phylogenetic analysis and neutralization tests [6, 7, 12]. The presence of EHDV-7 and IBAV within the same geographic region in Japan provides opportunities for these two different serotypes to infect the same animal simultaneously, with the potential to generate reassortment viruses.
To investigate the epidemiology and distribution of bovine insect-borne viruses, sentinel herds of cattle were established in Yunnan Province in China. Heparinized whole-blood samples were collected from sentinel cattle in 2013, and the separated blood cells were lysed for virus isolation. A virus, designated YN09-04, was successfully isolated after one passage in C6/36 cells followed by three passages in BHK-21 cells. YN09-04 was identified as EHDV by reverse transcription PCR targeting S7 (data not shown) and was then further analyzed by sequencing its genome. Sequence-independent cDNA synthesis and complete genome amplification were performed according to the method of full-length amplification of cDNAs [5, 8]. The genome of YN09-04 is 19,344 bp in size and consists of ten segments ranging from 810 bp (S10) to 3942 bp (S1) in length, and the nucleotide (nt) sequences of these segments have been deposited in the GenBank database under accession numbers MK656453 to MK656462. The general characteristics, including the length of each segment, noncoding regions (NCRs), regions encoding amino acid (aa), and terminal hexanucleotides, are shown in Table 1. Comparative pairwise analysis (CLUSTAL W) of the full-length genome of YN09-04 versus EHDV strains available in GenBank is shown in Table 2. Phylogenetic trees (MEGA-X) based on the full-length nt sequence of each segment were constructed using the maximum-likelihood method, Jukes-Cantor model, and 1,000 bootstrap replicates.
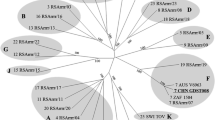
S2 and S6 encode the outer capsid proteins VP2 and VP5, respectively. Phylogenetic analysis of S2 revealed four distinct clusters (A to D) [3], which are correlated with the serological properties of the virus (Fig. 1a). YN09-04 grouped together with EHDV-2 and EHDV-7 isolates to form cluster A, in which the YN09-04 and EHDV-7 strains formed a distinct subclade that was completely separate from the EHDV-2 strains. Additionally, YN09-04 was found to be more closely related to Japanese EHDV-7 isolates than to those from Australia and Israel. Similarly, an S6-based phylogenetic tree also delineated four distinct groups (A to D) that closely resembled the pattern of S2 (Fig. 1b). Within cluster A, the YN09-04 S6 shares the highest nt and amino acid (aa) sequence similarity and the closest relationship with Japanese EHDV-7 isolates. VP2 is the major serotype-determining protein, while VP5 may influence the conformation of VP2 and exhibits some correlation with EHDV serotypes [3, 14]. The results indicate that YN09-04 belongs to EHDV serotype 7, and its S2 and S6 may share a common ancestor with Japanese EHDV-7 isolates.

Phylogenetic trees of S2 (a), S6 (b), S3 (c) and S7 (d) from YN09-04 and other EHDV strains with sequences available in the GenBank database. Only bootstrap values > 70% are shown at branch points. Chuzan virus was used as the outgroup to root the trees. The black triangle symbol indicates the Chinese isolate YN09-04
Segments 3 and 7 encode the inner capsid proteins VP3 and VP7, respectively. The YN09-04 S3 shares the highest sequence similarity with the Japanese strain Ibaraki BK13 at both the nt and aa level. A phylogenetic tree of S3 revealed that YN09-04 belongs to the eastern group (EHDVs isolated in Australia and Japan) and is closely related to the Australian strain CPR3961A and Japanese IBAVs, which, together with YN09-04, form a subgroup that is separate from other Australian EHDVs and Japanese EHDV-7 isolates (Fig. 1c). The YN09-04 S7 shows higher nt and aa sequence similarity to Japanese IBAVs than to other strains and therefore clusters with these IBAVs in the phylogenetic tree (Fig. 1d). These results demonstrate that the S3 and S7 of YN09-04 likely originated from EHDV strains similar to Japanese IBAV isolates.
The replicase complex is composed of VP1, VP4 and VP6, which are encoded by genome segments 1, 4 and 9, respectively. YN09-04 belongs to the eastern group based on phylogenetic analysis of S1, S4 and S9 (Fig. 2). YN09-04 S1 is closely related to Japanese IBAV isolates and shares the highest nt and aa identity with them (Fig. 2a). YN09-04 S4 shows maximum nt sequence similarity to the Australian strain CPR3961A, followed by Japanese IBAV isolates, with slightly less similarity but shares the highest aa sequence similarity with the Japanese strain Ibaraki. Thus, YN09-04 clusters with three strains, forming a subclade within the eastern group that is completely separate from other Australian EHDV isolates (Fig. 2b). Phylogenetic analysis of S9 showed that YN09-04 shares higher nt and aa sequence similarity with EHDV isolates from Australia than that with those from Japan. Furthermore, YN09-04 and Australian EHDV isolates cluster together and form a distinct subclade within the eastern group (Fig. 2c). These results indicate that the S1, S4 and S9 of YN09-04 apparently originated from eastern EHDV strains, and likely shares common ancestors with Australian EHDV and Japanese IBAV strains.

Phylogenetic trees of S1 (a), S4 (b), S9 (c), S5 (d), S8 (e) and S10 (f) from YN09-04 and other EHDV strains with sequences available in the GenBank database. Only bootstrap values > 70% are shown at branch points. Chuzan virus was used as the outgroup to root the trees. The black triangle symbol indicates the Chinese isolate YN09-04
The nonstructural proteins NS1, NS2 and NS3/NS3a are encoded by segments 5, 8 and 10, respectively. A phylogenetic tree of S5 shows that EHDV isolates can readily be divided into clear eastern and western (isolated in the Middle-East, Africa and North America) groups and that YN09-04 is included in eastern group and is most closely related to Japanese IBAV isolates, with the highest nt sequence similarity (Fig. 2d). YN09-04 and EHDV isolates from Australia cluster together based on the analysis of S8 and form a separate group from EHDV isolates from other countries (Fig. 2e). The YN09-04 S10 shares much higher nt and aa sequence similarity with the Australian strains CSIRO775 and CPR3961A than with other EHDV strains, and forms a separate cluster with these two Australian strains (Fig. 2f). These results demonstrate that the S5 of YN09-04 might share a common ancestor with those of Japanese IBAV strains, while its S8 and S10 likely share a common ancestor with those of Australian EHDV strains.
The YN09-04 S2 and S6 sequences are most similar to those of Japanese EHDV-7 strain KSB-14/E/97. Although YN09-04 has been identified as EHDV-7, the S1, S5 and S7 of YN09-04 are most similar to those of Japanese IBAV isolates, with which they cluster in the phylogenetic trees. Furthermore, the YN09-04 S3 and S4 grouped with Japanese IBAV isolates and Australian strain CPR3961A, forming a subclade that is completely separated from other Australian EHDVs within the eastern group. The EHDV S3 and S4 clustered strongly based on the origin of isolates (eastern/western, and were further divided into subclades that appear to reflect more local geographical groups [1]. Therefore, it is presumed that the S1, S3, S4, S5 and S7 of YN09-04 originated from EHDV strains that were similar to the Japanese isolates. In addition, the Chinese isolate YN09-04 and the Japanese IBAV isolates probably share ancestors that provided these genome segments. In addition, the phylogenetic trees of S8 and S9 showed that the YN09-04 and Australian EHDVs cluster together, reflecting their common origin. The YN09-04 S10 was most closely related to sequences from Australian strains CSIRO 775 and CPR 3961A and formed a distinct clade separated from other EHDVs. The EHDV S8 is clearly characterised by geographic clustering; the S9 sequences divide primarily into eastern and western groups, and their sub-clusters are based on more local geographic relationships. The S10 has two forms, and both appear to exist in the east and the west [1, 2]. Thus, the S8, S9 and S10 of YN09-04 are obviously related to those of Australian EHDV isolates and likely originated from the corresponding segments of these Australian EHDV strains or similar isolates. Based on the analysis described above, we presume that the Chinese isolate YN09-04 is a reassortant, and its genome segments probably share common ancestors with EHDV strains from both Japan and Australia.
The current study describes the isolation, identification and whole-genome phylogenetic analysis of the EHDV-7 strain YN09-04, which was first isolated in China. Prior to this study, EHDV-7 and IBAV strains had never been reported in China. Our data indicate that the genetic composition of Chinese EHDV-7 strain YN09-04 is complex, and it likely originated from a reassortment event between EHDV strains that are similar to Japanese and Australian EHDV isolates. Unfortunately, due to the lack of continual surveillance of EHDVs throughout China and complete genome sequencing of more EHDV strains, we cannot provide detailed information regarding when and where the EHDV isolate of a new serotype was involved or has undergone reassortment.
References
Anthony SJ, Maan N, Maan S, Sutton G, Attoui H, Mertens PP (2009) Genetic and phylogenetic analysis of the core proteins VP1, VP3, VP4, VP6 and VP7 of epizootic haemorrhagic disease virus (EHDV). Virus Res 145:187–199
Anthony SJ, Maan N, Maan S, Sutton G, Attoui H, Mertens PP (2009) Genetic and phylogenetic analysis of the non-structural proteins NS1, NS2 and NS3 of epizootic haemorrhagic disease virus (EHDV). Virus Res 145:211–219
Anthony SJ, Maan S, Maan N, Kgosana L, Bachanek-Bankowska K, Batten C, Darpel KE, Sutton G, Attoui H, Mertens PP (2009) Genetic and phylogenetic analysis of the outer-coat proteins VP2 and VP5 of epizootic haemorrhagic disease virus (EHDV): comparison of genetic and serological data to characterise the EHDV serogroup. Virus Res 145:200–210
Kedmi M, Van Straten M, Ezra E, Galon N, Klement E (2010) Assessment of the productivity effects associated with epizootic hemorrhagic disease in dairy herds. J Dairy Sci 93:2486–2495
Maan S, Rao S, Maan NS, Anthony SJ, Attoui H, Samuel AR, Mertens PP (2007) Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J Virol Methods 143:132–139
Ohashi S, Yoshida K, Watanabe Y, Tsuda T (1999) Identification and PCR-restriction fragment length polymorphism analysis of a variant of the Ibaraki virus from naturally infected cattle and aborted fetuses in Japan. J Clin Microbiol 37:3800–3803
Ohashi S, Yoshida K, Yanase T, Tsuda T (2002) Analysis of intratypic variation evident in an Ibaraki virus strain and its epizootic hemorrhagic disease virus serogroup. J Clin Microbiol 40:3684–3688
Potgieter AC, Page NA, Liebenberg J, Wright IM, Landt O, van Dijk AA (2009) Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J Gen Virol 90:1423–1432
Savini G, Afonso A, Mellor P, Aradaib I, Yadin H, Sanaa M, Wilson W, Monaco F, Domingo M (2011) Epizootic haemorrhagic disease. Res Vet Sci 91:1–17
Schirtzinger EE, Jasperson DC, Ostlund EN, Johnson DJ, Wilson WC (2018) Recent US bluetongue virus serotype 3 isolates found outside of Florida indicate evidence of reassortment with co-circulating endemic serotypes. J Gen Virol 99:157–168
Shaw AE, Ratinier M, Nunes SF, Nomikou K, Caporale M, Golder M, Allan K, Hamers C, Hudelet P, Zientara S, Breard E, Mertens P, Palmarini M (2013) Reassortment between two serologically unrelated bluetongue virus strains is flexible and can involve any genome segment. J Virol 87:543–557
Shirafuji H, Kato T, Yamakawa M, Tanaka T, Minemori Y, Yanase T (2017) Characterization of genome segments 2, 3 and 6 of epizootic hemorrhagic disease virus strains isolated in Japan in 1985–2013: identification of their serotypes and geographical genetic types. Infect Genet Evol 53:38–46
Yadin H, Brenner J, Bumbrov V, Oved Z, Stram Y, Klement E, Perl S, Anthony S, Maan S, Batten C, Mertens PP (2008) Epizootic haemorrhagic disease virus type 7 infection in cattle in Israel. Vet Rec 162:53–56
Zhang X, Boyce M, Bhattacharya B, Zhang X, Schein S, Roy P, Zhou ZH (2010) Bluetongue virus coat protein VP2 contains sialic acid-binding domains, and VP5 resembles enveloped virus fusion proteins. Proc Natl Acad Sci USA 107:6292–6297
Acknowledgements
Funding
This study was supported by Special Fund for Agro-Scientific Research in the Public Interest of China (No. 201303035).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors have conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Tim Skern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Qi, Y., Wang, F., Chang, J. et al. Identification and complete-genome phylogenetic analysis of an epizootic hemorrhagic disease virus serotype 7 strain isolated in China. Arch Virol 164, 3121–3126 (2019). https://doi.org/10.1007/s00705-019-04412-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04412-9