
Abstract
A new virus with a circular double-stranded DNA genome was discovered in green Sichuan pepper with vein clearing symptoms. Its complete genome of 8,014 bp contains three open reading frames (ORF) on the plus strand, which is typical of members of the genus Badnavirus in the family Caulimoviridae. Sequence comparisons revealed that the new virus has the highest nucleotide sequence identity with grapevine vein-clearing virus (GVCV). In particular, the identity of the two viruses in the ORF3 RT-RNase H region is 71.9%, which is below the species demarcation cutoff of 80% for badnaviruses. Phylogenetic analysis also placed the new virus with GVCV in a cluster. The virus was tentatively named “green Sichuan pepper vein clearing-associated virus” (GSPVCaV). The geographical distribution and genetic diversity of GSPVCaV were studied. Another isolate was found to be highly divergent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Green Sichuan pepper or Chinese pepper (Zanthoxylum schinifolium, Rutaceae) has been cultivated for the production of fruits and leaves in China for a long time. Its essential oil extract is known to have medicinal activities [1]. The extract also contains aromatic substances, so it is widely used as a spice in China and other East Asian countries. Several pathogens, including fungi and phytoplasma, have been described in the crop, causing serious damage [2, 3]. However, no viral disease has been reported in green Sichuan pepper.
Members of the genus Badnavirus in the family Caulimoviridae are important agricultural and horticultural pathogens that cause serious crop losses [4]. These viruses have non-enveloped, bacilliform virions of 120–150 × 30 nm and a single circular double-stranded DNA of 7.2–9.2 kb that contains three open reading frames (ORFs) arranged in a series on the plus strand [4]. In recent years, several new badnaviruses have been identified in various crops [5,6,7,8]. In 2018, virus-like vein clearing and mottling symptoms (Fig. 1a–c) were observed on the leaves of green Sichuan pepper trees in Chongqing Province, China. Total nucleic acids were extracted from leaf tissues of the symptomatic tree using an EASYspin Plus Complex Plant RNA Kit (Aidlab, China). An rRNA-depleted cDNA library was built and subjected to Illumina NextSeq sequencing with a layout of 150-bp paired-end (PE) reads (Biomarker Technologies, Beijing, China). Analysis of total sequenced reads was performed using CLC Genomics Workbench 9.5 (QIAGEN, USA) as described by Shen et al. [9], with sequences of the adaptors and reads of low quality and host sequences removed. The remaining reads were assembled de novo into contigs, and BLAST search programs were used for contig annotation (Supplementary Table S1). To determine the complete viral genome sequence, overlapping primer pairs (Supplementary Table S2) were designed based on the viral contigs. Total nucleic acids obtained from symptomatic leaves using a DNAsecure Plant Kit (Tiangen Biotech, China) were used for PCR amplification. PCR products were purified and cloned into the pEASY-T1 vector (TransGen Biotech, China). At least five clones for each amplicon were sequenced, and the sequences that were obtained were assembled using SeqMan software (DNAStar Lasergene, USA). ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), Conserved Domain Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), and DRNApred (http://biomine.cs.vcu.edu/servers/DRNApred/) were used to predict the genomic ORFs, identify conserved protein domains, and predict DNA/RNA binding sites, respectively.

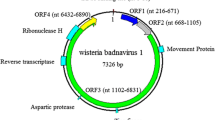
Viral symptoms on a twig (a), a young leaf (b), and mature leaf (c) associated with green Sichuan pepper vein clearing-associated virus (GSPVCaV) in a green Sichuan pepper tree. (d) Schematic genome organization of GSPVCaV showing three open reading frames (ORF1–ORF3) as well as the tRNA primer binding site and conserved functional motifs in viral proteins
A total of 55,749,940 clean reads were obtained, and 17,782,807 (31.9%) of them were used for BLASTx annotation after removal of the reads mapped to the referenced Citrus (Rutaceae) genomes, as no genome sequences were available for Zanthoxylum species. De novo assembly of unmapped reads produced 87,834 contigs. One contig of 7,994 nt showed significant nucleotide (nt) sequence identity (56.5%) to grapevine vein-clearing virus (GVCV), suggesting the presence of a new badnavirus in the sample. Mapping of transcriptome reads as well as RT-PCR detection of viral mRNAs (Supplementary Fig. S1 and Supplementary Table S2) suggested that this is an episomal virus rather than fragments integrated into the genome of the host plant. The virus was provisionally named “green Sichuan pepper vein clearing-associated virus” (GSPVCaV, isolate CQ-1), as it was the sole virus found by sequencing the cDNA library from the symptomatic tree.
The complete genome of GSPVCaV is 8,014 nt in size (GenBank accession No. MK371353). Comparisons with other badnaviruses identified a putative tRNAmet-binding site (5′-1TGGTATCAGAGCTTCGGC18), a potential 7,xxxTATA7,xxx+4 box, and a possible polyadenylation signal (7,xxxAATAAA7,xxx+6) within the intergenic region on the plus strand [10]. The genome of the virus is typical of badnaviruses, containing three open reading frames (ORF) (Fig. 1d). ORF1 (nt 359–982) encodes a putative protein (P1, 23.7 kDa) of 207 amino acid (aa) residues that contains a motif (pfam07028, aa 88–189) that is conserved among badnaviruses [8]. The P1 of unknown function has been associated with virions [11]. ORF2 (nt 979–1,368) is predicted to encode a 129-aa protein (P2, 14.4 kDa) with a DNA-binding site at aa 49–129, suggesting non-specific DNA- and RNA-binding activities [12]. ORF3 (nt 1,365–7,538) encodes a large polyprotein of 2,057 aa (P3, 232.5 kDa). Like other badnaviruses, the polyprotein contains the coat protein, movement protein (MP), a highly conserved zinc-finger-like RNA-binding domain (CXCX2CX4HX4C), reverse transcriptase (RT), viral aspartic protease (AP), and ribonuclease H (RNase H) domains [13]. Genome-wide comparisons with badnaviruses confirmed that GSPVCaV is most closely related to GVCV. The sequence identities between the two viruses are 56.5% for the whole genome nt sequence, and 64%, 51.2% and 56.0% for the P1, P2 and P3 aa sequences, respectively. The nt sequence identity of the joint region of the RT and RNase H domains of P3 is 71.9%, which is lower than the species demarcation cutoff (80%) for the genus Badnavirus [4], suggesting that GSPVCaV should be considered a member of a new species in this genus.
A phylogenetic tree was generated by the neighbour-joining method from the alignment of the genome sequences of GSPVCaV and representative badnavirus sequences retrieved from the GenBank database. The phylogenetic analysis placed GSPVCaV and GVCV in the same cluster, confirming their close relationship and supporting that GSPVCaV is a distinct badnavirus (Fig. 2).

Neighbor-joining phylogenetic tree derived from an alignment of the complete genome sequences of GSPVCaV and other extant badnaviruses, with 1000 bootstrap replicates. Bootstrap values are shown at the relevant nodes, and only bootstrap values higher than 50% shown. The scale bar represents substitutions per site
Our field surveys showed that vein clearing and similar symptoms were common in green Sichuan pepper in Sichuan Province. Another leaf sample (SC-1) was subsequently collected from trees at different locations and subjected to Illumina NextSeq sequencing. Bioinformatics analysis identified only one viral contigs with high sequence similarity to GSPVCaV-CQ1. The complete genome sequence of 8,070 nt (MK371354) was obtained by Sanger sequencing for this isolate. The SC-1 isolate is 94.0% identical to the CQ1 isolate. Sequence differences mainly concentrated at the NCR (7,525–359 nt), with the exception of a 15-nt insertion at nt 4,669. A virus-specific primer pair (HJV-F/-R; Table S2) was designed based on the RT-RNaseH region for the two isolates and used in PCR to test 25 symptomatic and six asymptomatic trees. The virus was detected in 23 of the 25 symptomatic trees (92.0%) but not in any asymptomatic trees (data no shown), suggesting a tight association of the virus with the vein clearing symptoms. However, the biological characteristics of the isolates CQ-1 and SC-1 and the biological differences between them remain undetermined, as Koch’s postulates are yet to be fulfilled.
References
Li W, Zhou W, Shim SH, Kim YH (2014) Chemical constituents of Zanthoxylum schinifolium (Rutaceae). Biochem Syst Ecol 55:60–65
Kirk PM, Cannon PF, Minter DW, Stalpers JA (2008) Ainsworth & Bisby’s dictionary of the fungi, edition, 10th edn. CAB International, Wallingford
Lu YJ, Li GL (1995) A new species of the genus Marssonina. Acta Mycol Sin 14:184–186
Geering ADW, Hull R (2012) Family caulimoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, pp 429–443
James AP, Geijskes RJ, Dale JL, Harding RM (2011) Molecular characterisation of six badnavirus species associated with leaf streak disease of banana in East Africa. Ann Appl Biol 158:346–353
Maliogka VI, Olmos A, Pappi PG, Lotos L, Efthimiou K, Grammatikaki G, Candresse T, Katis NI, Avgelis AD (2015) A novel grapevine badnavirus is associated with the Roditis leaf discoloration disease. Virus Res 203:47–55
Kazmi SA, Yang Z, Hong N, Wang G, Wang Y (2015) Characterization by small RNA sequencing of taro bacilliform CH virus (TaBCHV), a novel badnavirus. PLoS One 10:e0134147
Xu D, Mock R, Kinard G, Li R (2011) Molecular analysis of the complete genome of four isolates of Gooseberry vein banding associated virus. Virus Genes 43:130–137
Shen P, Tian X, Zhang S, Ren F, Li P, Yu YQ, Li RH, Zhou CY, Cao MJ (2018) Molecular characterization of a novel luteovirus infecting apple by next-generation sequencing. Arch Virol 163:761–765
Bouhida M, Lockhart BEL, Olszewski NE (1993) An analysis of the complete sequence of a sugarcane bacilliform virus genome infectious to banana and rice. J Gen Virol 74:15–22
Cheng CP, Lockhart BEL, Olszewski NE (1996) The ORFI and II proteins of Commelina yellow mottle virus are virion-associated. J Gen Virol 223:263–271
Jacquot E, Hagen LS, Jacquemond M, Yot P (1996) The open reading frame 2 product of Cacao swollen shoot badnavirus is a nucleic acid-binding protein. Virology 225:191–195
Hohn T, Fütterer J, Hull R (1997) The proteins and functions of plant pararetroviruses: knowns and unknowns. Crit Rev Plant Sci 16:133–161
Acknowledgements
This research was supported by the Intergovernmental International Science, Technology and Innovation (STI) Collaboration Key Project of China’s National Key R&D Programme (NKP) (2017YFE0110900), Fundamental Research Funds for the Central Universities (XDJK2018AA002), Chongqing Research Program of Basic Research and Frontier Technology (cstc2017jcyjBX0016), and Overseas Expertise Introduction Project for Discipline Innovation (111 Center) (B18044). We thank the editor and two anonymous reviewers for their constructive comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
I have read and abided by the statement of ethical standards for manuscripts submitted to Archives of Virology.
Conflict of interest
All authors declare they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: F. Murilo Zerbini.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Min Xu and Song Zhang are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Detection of viral mRNAs by RT-PCR and HTS (transcriptome read mapping). M, DL2000 DNA marker; 1 and 2, ORF1; 3 and 4, ORF2; 4 and 5, ORF3 (JPEG 182 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Xu, M., Zhang, S., Xuan, Z. et al. Molecular characterization of a new badnavirus infecting green Sichuan pepper (Zanthoxylum schinifolium). Arch Virol 164, 2613–2616 (2019). https://doi.org/10.1007/s00705-019-04357-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04357-z