
Abstract
Species A rotavirus still remains a major cause of acute gastroenteritis in infants and young children. Globally, six genotypes (G1P[8], G2P[4], G3P[8], G4P[8], G9P[8] and G12P[8]) account for >90% of circulating strains; however, genotype G12 in combination with P[6] or P[9] has been detected at increasing rates. We sought to broaden our knowledge about the rotavirus strains circulating during the early post-vaccine-introduction period. Stool samples were obtained from children hospitalised for acute gastroenteritis in Belém, Northern Brazil, from May 2008 to May 2011 and examined by reverse transcription polymerase chain reaction and nucleotide sequencing. A total of 122 out of the original 1076 rotavirus strains were judged to be non-typeable in the first analysis and were therefore re-examined. G2P[4] was the most prevalent genotype (58.0%), followed by G1P[8] (16.9%), and G12P[6] (7.5%). G12P[6] strains were identified at similar rates during the first (2.5%) and second (3.9%) years, and the rate jumped to 15.6% in the third year. Analysis of VP7 sequences of the G12P[6] strains showed that they belonged to lineage III. In addition, co-circulating G12P[6] strains displaying long and short RNA patterns were found to belong to the Wa-like and DS-1-like constellation, respectively. Additional unusual circulating strains G12P[9] and G3P[9] were also identified. This hospital-based study showed a high prevalence of G12P[6] strains in the third year of surveillance. Our results highlight the need for continuous longitudinal monitoring of circulating rotavirus strains after introduction of rotavirus vaccines in Brazil and elsewhere.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the introduction of rotavirus vaccination into the national immunisation programs (NIPs) of almost 100 countries, group A rotaviruses (RVA) still remain a leading cause of deaths and hospitalisations due to gastroenteritis (GE) among children aged <5 years all over the world. Recently, the Global Burden of Diseases Study, 2015 [1] confirmed RVA to be the most common cause of diarrhoea in children younger than age 5 years, causing an estimated 259,700 (211,200-323,500) deaths per year in 2015. Prior to the widespread use of rotavirus vaccines in Brazil, RVA infection was responsible for approximately 650,000 outpatient visits, 92,000 hospitalisations, and 850 deaths annually in children under five years of age [2]. Worldwide, Brazil was among the first and largest countries to introduce a rotavirus vaccine into the public sector, covering a birth cohort of approximately 3 million infants [3]. In March 2006, the live-attenuated monovalent human-derived G1P[8] rotavirus vaccine Rotarix® (GlaxoSmithKline Vaccines, Rixensart, Belgium) was incorporated into the country’s national immunisation program with the aim of reaching 90% coverage (2 doses) among infants aged two to four months [4]. Twelve years have elapsed since the implementation of rotavirus vaccination into the public sector in Brazil, and a considerable amount of evidence has accumulated showing a substantial reduction in GE-related hospitalisations and deaths, particularly among children aged less than one year [4,5,6,7,8,9]. Of public-health importance was the fact that most of these studies also showed a decline in hospitalisations for GE among children too old to receive the rotavirus vaccine, suggesting a herd protection effect.
Rotaviruses constitute a genus within the family Reoviridae. Their genome consists of 11 segments of double-stranded RNA, which are surrounded by a non-enveloped, icosahedral triple-layered capsid [10, 11]. RVA possesses two outer layer proteins, named VP7 and VP4, which define the G (glycoprotein) and P (protease-sensitive) genotype, respectively. A total of 36 G and 51 P genotypes have been differentiated to date, of which at least 12 G types and 15 P types are commonly associated with infection in humans [11, 12]. Although over 60 G-P dual combinations have been reported in humans, five (G1P[8], G2P[4], G3P[8], G4P[8] and G9P[8]) are known to account for greater than 80% of the circulating genotypes causing childhood rotavirus gastroenteritis globally [13, 14]. Rotavirus strains with G12 genotype specificity were first detected in the Philippines in 1987 [15], but in the last decade, their prevalence has increased exponentially on a global scale, namely in combination with genotype P[6] or P[8]. Therefore, G12 is now recognised as the sixth epidemiologically important genotype associated with infections in humans [13, 16, 17]. Interestingly, genotype G3 has been reported to infect not only humans but also a broad range of other host species, and some uncommon G/P combinations, such as G3P[3] and G3P[9], are believed to be of canine, feline or human origin [18,19,20].
A comprehensive classification system has been proposed for RVA that is based on the whole viral genome and assigns a genotype to each of the 11 gene segments, as differentiated on the basis of specific cutoff points of nucleotide sequences [10, 21]. In this system, the abbreviation Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx is used to define the genotypes of the VP7, VP4, VP6, VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4, and NSP5/6 genes, of which at least 36 G, 51 P, 26 I, 22 R, 20 C, 20 M, 31 A, 22 N, 22 T, 27 E, and 22 H genotypes has been identified in humans and/or animals [12, 22].
Although Brazil is likely to be approaching the established target of 90% rotavirus vaccination coverage, at least for some regions, rotaviruses still contribute significantly to the burden of diarrhoea in children aged <5 years, particularly in the impoverished Northern and Northeast regions [4]. Moreover, the nationwide implementation of rotavirus vaccination in Brazil in 2006 raised some concern that vaccine-induced selective pressure might have occurred, since a considerable increase in the prevalence of G2P[4] rotavirus strains was observed throughout the country, particularly during 2007-2009 [4, 23, 24]. However, more-recent surveillance studies conducted in Brazil have shown a sharp decline in the prevalence of G2P[4] genotypes followed by an increase in the occurrence of G1P[8] and others such as G12P[8], supporting the hypothesis of natural fluctuation of strains temporally and regionally, rather than a replacement mechanism [25,26,27,28].
In a previous, 3-year hospital-based monitoring study of rotavirus strains conducted in Belém, Northern Brazil, we focussed our VP7 characterisation only on the genotypes that are most prevalent worldwide which have G1, G2, G3, G4, or G9 type specificity [26, 29]. In the present study, we sought mainly to provide additional information about the circulating rotavirus strains during the early post-vaccine-introduction period in our region, including those of G and P genotypes that are currently recognised as emerging or unusual and that may eventually pose a challenge to current vaccination strategies.
Materials and methods
From May 2008 to May 2011, a hospital-based study was carried out in Belem, Northern Brazil, with two main objectives. First, to determine the effectiveness of the monovalent G1P[8] human rotavirus vaccine (Rotarix®) between May 2008 and May 2009, and second, to monitor the occurrence of circulating rotavirus strains throughout the whole study period (May 2008 to May 2011). A full description of the methods and results from these original studies were provided elsewhere, including the genotype characteristics of the viruses from 1076 rotavirus-positive samples, using G or P oligonucleotide primers targeting the G (G1, G2, G3, G4, and G9) and P (P[4], P[6], P[8], and P[9]) RVA types [26, 29]. Of these, 122 (11.3%) strains could not be fully G- and/or P-typed, and a further attempt to genotype them by including a primer for G12 was the main objective of the current study.
The study protocol was approved by the IRB Committee of the Brazilian Ministry of Health’s National Rotavirus Reference Laboratory at Instituto Evandro Chagas (IEC), under reference number 579.295, and the study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Written informed consent was obtained from the parents/legal guardians before enrolment.
Stool samples were prepared as 10% v/v suspension in 0.01 M Tris-Ca++, pH 7.2, and viral double-stranded (ds) RNA was extracted from the faecal supernatant using guanidinium isothiocyanate-silica as described by Boom et al. [30]. To determine electropherotypes, the extracted dsRNAs were further electrophoresed in a 5% polyacrylamide gel, followed by silver staining as described by Pereira et al. [31].
Reverse transcription polymerase chain reaction (RT-PCR) of the VP4 and VP7 genes was done for all strains, using the consensus primers 4Con3-4Con2 and Beg9-End9, respectively, as described previously [32, 33]. The G and P genotyping was performed in a second-round RT-PCR (hemi-nested PCR) using different specific primers targeting G5, G6, G8, G10 e G12 and P[1], P[5], P[7], P[10] and P[11] [32, 34].
Nucleotide sequencing was carried out with isolates that could not be genotyped previously by RT-PCR. In brief, gel-purified first-round RT-PCR amplicons from the VP7 and VP4 genes were sequenced using a BigDye Terminator Cycle Sequencing Reaction Kit v3.1 (Applied Biosystems, Foster City, CA) on an ABI Prism 3130xl Genetic Analyser (Applied Biosystems).
Sequences were analysed, and consensus sequences were prepared using the CAP3 sequence assembly program. Multiple consensus alignments were made using the software MAFFT v.7.221 [35], and sequence editing was done using the Geneious Bioinformatics software platform v.8.1.7 [36]. The data were compared with the corresponding sequences from the National Center for Biotechnology Information GenBank database using the BLAST alignment tool [37].
Molecular phylogenetic analysis was done using the IQ-TREE v.1.3.2 package for inferring maximum-likelihood trees [38]. Trees were drawn to scale with branch lengths measured in the number of substitutions per site, and the statistical significance was assessed by bootstrap resampling analysis (1000 pseudoreplicates). The resulting phylogenetic trees were visualized using the program FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). The sequences obtained in this study have been deposited in the GenBank database under the accession numbers MF695027-MF695065.
We further selected two G12P[6] strains with identifiable RNA profiles (2A1194 and 2A3406 with long and short electropherotypes, respectively) to perform a whole-genome analysis using specific primers for amplification of the VP1 (686 bp), VP2 (686 bp), VP3 (702 bp), VP6 (1356 bp), NSP1 (1,565 bp), NSP2 (1,038 bp), NSP3 (1,062 bp), NSP4 (738 bp), and NSP5 (664 bp) genes as reported before [39,40,41,42]. Amplicons were subjected to nucleotide sequencing, essentially as described above, and a partial nucleotide sequences for each gene segment was determined and deposited in the GenBank database under the following accession numbers: KX279732.1 (VP6), KX279702.1 (VP3), KX274667.1 (VP2), KX274608.1 (NSP5), KX274578.1 (NSP4), KX274548.1 (NSP3), KX274518.1 (NSP2), and KX274488.1 (NSP1) for the Wa-like strain 2A1194; and KX279740.1 (VP6), KX279710.1 (VP3), KX274675.1 (VP2), KX274645.1 (VP1), KX274616.1 (NSP5), KX274586.1 (NSP4), KX274556.1 (NSP3), KX274526.1 (NSP2), and KX274496.1 (NSP1) for the DS1-like strain 2A3406.
Statistical analysis was performed using SAS version 9.1 (SAS Institute Inc., Cary, NC). The statistical significance of differences between prevalence rates of genotypes over time was evaluated by the χ2-test or Fisher’s exact test (whenever applied). All p-values were two-tailed, and p < 0.05 was considered to be statistically significant.
Results
From May 2008 to May 2011, there were a total of 122 (11.3%) out of the original 1076 RVA strains that could not be partially or fully typed in a previous assessment of circulating genotypes in Belém, Brazil, because the set of primers used did not target G12 strains [26]. These strains were thus reexamined using either a broader array of primers or nucleotide sequencing. Overall (combining the previous and current genotyping assessments), G2P[4] isolates were predominant, accounting for 58.0% (625/1076) of the genotyped samples (Table 1). The second and third most prevalent strains were G1P[8] and G12P[6], representing 16.9 (182/1076) and 7.5% (81/1076) of the genotyped strains, respectively.
Among the remaining dual G and P combinations, mixed and partially or fully untypeable RVA strains represented 10.9% (117/1076) and 1.5% (16/1076) of the total samples, respectively. In addition, the globally emerging G12P[8] and the rare G12P[9] and G3P[9] genotypes were found among typed strains at rates of 0.6% (6/1076), 0.4% (4/1076) and 0.1% (1/1076), respectively.
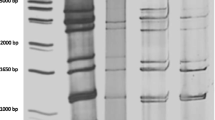
Fig. 1 shows the electropherotypes of six representative G12P[6] rotavirus strains, of which four displayed identical long patterns with a 4-2-3-2 distribution of the RNA segments, whereas two others had a typical short electrophoretic profile also exhibiting clustering of RNA fragments into a 4-2-3-2 configuration.

Representative electrophoretic profiles of G12P[6] rotavirus strains (lanes 1-4, long RNA profiles; lanes 5-6, short RNA profiles) detected in Belém, Brazil, and controls (C1, G1P[8], long RNA profile; C2, G2P[4], short RNA profile)
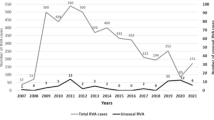
The distribution of RVA genotypes throughout the 37-month study is shown in Fig. 2. G12P[6] genotypes were identified at similar rates during the first (2.5%; 13/525) and second (3.9%; 10/256) years, and this rate jumped to 15.6% (46/295) in the third year. G2P[4] strains were found to be predominant from May 2008 to February 2010, occurring at rates that ranged from 61% to 100%. From March 2010 onwards, the prevalence rate of the G2P[4] genotype declined sharply, reaching proportions as low as 8% in December 2010. Whilst the G1P[8] genotype was not identified from May 2008 to January 2009, an increase in G1P[8] prevalence was observed from April 2010 to May 2011, yielding rates in the range of 36% to 79% of circulating strains.

Annual distribution of rotavirus genotype G and P combinations in Belém, Brazil, from May 2008 to May 2011. Others, untyped strains; mixed, mixed G and/or P types
The rest of the isolates included a variety of uncommon rotaviruses, as well as mixed and partially or fully untypeable strains, which were in general circulating at rates <10%.
The results of analysis of the VP7 sequences of G12P[6] (n = 17), G12P[8] (n = 4) and G12P[9] (n = 4) strains are depicted in Fig. 3a. Brazilian G12P[6] and G12P[8] strains clustered within lineage III, together with recent isolates from Korea, Slovenia, the USA, Bangladesh, Italy, and Botswana with nucleotide sequence identity among these strains ranging from 95.2% to 100%. When compared with other G12 strains used as references (the first G12 strains detected in our setting), the median nucleotide sequence identity was 98.8% and 96.4% for G12P[6] and G12P[8], respectively. G12P[6] strains displaying short (n = 8) and long (n = 4) electropherotypes were found to co-circulate during the study period. A high degree of genetic relatedness (98.9%-100% sequence identity) was observed among the four Brazilian G12 strains associated with the P[9] genotype, all of which clustered in lineage II, whereas less sequence similarity (<90% identity) was seen between these strains and other G12P[9] reference strains from Brazil, Argentina and Paraguay. Nucleotide sequence analysis of P[6] genotype associated with G12 revealed that all ten strains clustered in lineage I, and their sequences were 99.1% to 100% identical (Fig. 3b). Compared with other intra-lineage reference strains, nucleotide sequence identity rates ranged from 98.2% to 100%. Results from the analysis of VP4 sequences of the P[9] genotypes, including four G12P[9] strains and one G3P[9] strain, are summarized in Fig. 3c. All Brazilian G12P[9] strains clustered in lineage II, whereas the G3P[9] strain (1A3739/2011) clustered with strains of lineage III. It was shown in our study that P[9] strains with G12-type specificity exhibited high nucleotide sequence similarity to each other (99.6%-100% identity), whereas the P[9] strain bearing G3-type specificity showed low genetic relatedness with both lineage I (86% to 88% identity) and lineage II (86.4% to 87.8% identity).


(a) Phylogenetic dendrogram based on VP7 gene sequences of local G12P[6], G12P[8] and G12P[9] strains (highlighted in shaded areas) detected during May 2008-May 2011 in Belém, Brazil. Closed triangles and circles represent G12P[6] strains displaying short and long RNA profiles, respectively. Bootstrap values based on 1000 replicates are shown. Intra-genotypic lineages are indicated in square brackets at the right. (b) Phylogenetic dendrogram based on VP4 gene sequences of local G12P[6] strains (highlighted in shaded areas) detected during May 2008-May 2011 in Belém, Brazil. Bootstrap values based on 1000 replicates are shown. Intra-genotypic lineages are indicated in square brackets at the right. (c) Phylogenetic dendrogram based on VP4 gene sequences of local G12P[9] strains (highlighted in shaded areas) detected during May 2008-May 2011 in Belém, Brazil. Bootstrap values based on 1000 replicates are shown. Intra-genotypic lineages are indicated in square brackets at the right
A full genomic characterisation of two G12P[6] rotaviruses demonstrated that while strain 2A1194 (with a long RNA profile) possessed a Wa-like (I1-RX-C1-M1-A1-N1-T1-E1-H1) genotype constellation, strain 2A3406 (short RNA profile) possessed a DS-1-like (I2-R2-C2-M2-A2-N2-T2-E2-H2) genotype constellation.
Discussion
The present analysis represents an extension of a previous three-year surveillance follow-up of RVA strains in Belém, Brazil, circulating within five years after introduction of RVA vaccination into the Brazilian national immunisation program [26]. It was observed during the previously published study that a high proportion of strains remained untypeable for the G and/or P type, most probably due to the fact that we used a set of primers that did not target G12, which was then known to be a globally emerging genotype, and there had been a previous report on the detection of G12P[6] strains in the Northern region of Brazil [43]. In addition, nucleotide sequencing could not be performed with the strains that were previously untypeable by RT-PCR, suggesting that we might have missed G12 strains bearing either P[6] or P[8] types. In order to improve our knowledge of circulating rotaviruses in the post-vaccination period in our region, we reassessed VP7 and VP4 genotypes in the subset of strains that had not typed previously, using a set of primers targeting a broader range of genotypes.
Of particular interest in our study was the observation that G12 strains accounted for 10% of the isolates; of these, >80% were found to possess P[6] genotype specificity. G12P[6] strains were identified at rates ranging from 2.5% to 4% during the first and second years, and this rate jumped to 15.6% in the third year. This trend toward a higher frequency during the third year of follow-up is consistent with recent findings showing that G12 genotype possessing either P[6] or P[8] specificity became predominant in several countries throughout the world [13, 44]. Moreover, the proportion of circulating G12P[6] strains detected in our study appeared to be in line with the rates that were first reported in the Brazilian Amazon region [42, 45] and elsewhere [27, 28, 46,47,48,49,50], which were within the range of 1% to 17%. In this context, specific data from Brazil suggested that a G12P[6] “season” might have occurred in our region in 2010/2011.
A finding of particular interest in our study was the detection of co-circulating G12P[6] strains with long and short RNA patterns, raising the question as to whether they might belong to distinct genotype constellations. We were able to carry out a whole-genome characterization of two representative G12P[6] isolates found that that strains with long and short RNA migration patterns belonged to the Wa-like and DS1-like genotype constellation, respectively. This might eventually demonstrate that, in our surveillance study, G12P[6] rotaviruses evolved in Belém, Brazil, through the occurrence of reassortment events between locally co-circulating strains. Likewise, Nakagomi et al. [49] have reported the co-circulation of G12P[6] rotaviruses in Blantyre, Malawi, between 2007 and 2008, where strains possessing a long RNA profile displayed a complete Wa-like genotype constellation, whereas those with a short RNA pattern were found to be single VP3 gene substitution reassortants with a DS-1-like genetic backbone. Additional research at both the nucleotide and amino acid level should be conducted with these distinct G12P[6] strains in Belém for a better understanding of their evolutionary dynamics in our region.
Interestingly, phylogenetic analysis of VP7 gene sequences of all of our G12P[6] strains showed that they clustered in lineage III, which is known to include the majority of the G12 strains bearing either P[6] or P[8] genotype specificities in Brazil and elsewhere [17, 43, 44]. Of note in this context, data from the current study confirm previous findings in northern Brazil by Soares et al. [43], who demonstrated that the VP7 gene of G12P[6] rotavirus strains also belonged to lineage III. Altogether, these results show that a broader phylogenetic analysis of G12 strains that have emerged and spread across the Amazon during the past ten years is warranted and would provide a better understanding of their evolutionary genetics.
Since several pre- and post-rotavirus-vaccine-introduction studies have shown that the two licensed vaccines (Rotarix® and RotaTeq®) provide protection against a broad variety of RVA strains, including those not incorporated in these formulations, it seems improbable that the emergence of G12P[6] in our region will pose a challenge to the current vaccination strategies [29, 51, 52]. Furthermore, it should be pointed out that studies in Africa have shown that Rotarix® provides protection against diverse circulating rotavirus strains, including genotypes bearing P[6] and G12 specificities [53].
The occurrence of uncommon genotypes in our study also included the detection at low rates of five rotavirus strains bearing P[9] type specificity, combined with G12 (four strains) or G3 (one strain). Our results showing that all VP7 sequences from G12P[9] rotaviruses clustered together with lineage II of the VP4 gene are consistent with those reported previously in Korea, Japan, Argentina, Brazil and Paraguay [54,55,56,57]. In addition, the fact that P[9] is a highly prevalent genotype in the feline population raises the hypothesis of interspecies transmission, which could have generated the emerging G12P[9] strain in the Northern region [17, 58]. In order to determine the true origin of these strains, however, further sequencing studies are required, including the whole-genome characterisation of G12P[9] strains. In our study, there was a single isolate of the G3P[9] genotype of human rotavirus, which accounted for only 0.8% of all rotavirus-related gastroenteritis cases. Worldwide, the occurrence of P[9] strains has been associated with sporadic cases of gastroenteritis in humans, in general representing <2.5% of cases, even though significantly higher prevalence rates have been reported in Brazil (10.2%) and Poland (67.2%) [59,60,61]. In line with our findings, the majority of the P[9] genotypes that have been isolated elsewhere have had G1, G2, or G3 VP7-type specificity, and specifically the unusual G3P[9] combinations have been proposed to have been generated through reassortment events involving human and animal strains [62].
Interestingly, phylogenetic analysis revealed that G3P[9] strain 1A3739/2011 has a distant relationship to the prototype AU-1-like G3P[9] strain discovered in Japan in 1982, which is known to be closely related to a feline rotavirus strain, suggesting a possible zoonotic transmission from a cat to a human [63, 64]. Notably, our G3P[9] strain 1A3739/2011 clustered with another local G3P[9] strain (QUI-35-F5/2010), which was detected in an African-descendant community in Pará state, Brazil (J D P Mascarenhas, personal communication). Altogether, these findings suggest the emergence of a novel genetic variant of the P[9] genotype, possibly representing a new lineage. Surprisingly, previous molecular characterisation of the 1A3739/2011 strain in our setting revealed that it does not have an AU-like genotype constellation but rather possesses a multi-reassortant nature, with genes derived from chiropters, alpacas, equines, and simians [65].
In addition to expanding our observations on the occurrence of circulating G12P[6] strains in our setting, we identified common G1P[8] and G2P[4] genotypes at prevalence rates of 14.7% and 9.2%, respectively, among samples that had been characterised as untypeable when analysed previously [26]. While we currently do not have a plausible explanation for this, it seems likely that technical factors might have accounted for these discrepant results. It is worth mentioning in this regard that a number of studies have shown a high failure rate of primer-based genotyping, which is often attributed to single-nucleotide polymorphisms at primer-binding sites, resulting in inconsistent results [66, 67]. Moreover, Iturriza-Gomara et al. [68] have shown that amino acid substitutions at positions 87 (Ala to Thr) and 96 (Asp to Asn) within the VP7 gene of G2 rotavirus strains may be associated with failure to obtain a genotype determination. Further analysis will be performed with our local strains to assess whether possible failure of genotyping primers may have occurred due to changes in the primer-binding regions.
We found one genotype, G3P[8], that is commonly detected worldwide in humans, but further genomic characterisation was not performed to assess whether it would eventually represent a newly emergent reassortant equine-like strain, as reported in several recent studies in Brazil and elsewhere [69,70,71,72,73,74,75].
A major limitation in this study was the lack of a full analysis of the complete RVA genome constellations, which would allow a better understanding of the evolutionary dynamics of G12P[6] and G3P[9] strains spreading in our region. Furthermore, analysis of complete genome sequences would expand our knowledge on the genetic relationships between rotavirus strains of both human and animal origins.
In the context of the broad diversity of rotavirus strains circulating worldwide, this study strengthens the notion that long-term, continuous rotavirus surveillance will be essential to further clarify the overall impact of rotavirus vaccination. Our findings also provide additional evidence that simultaneous monitoring of rotavirus strains in humans and animals will lead to a better understanding of rotavirus ecology.
References
GBD Diarrhoeal Collaborators (2015) Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis of the Global Burden of Disease Study 2015. Lancet Infect Dis 17:909–948
Sartori AMC, Valentim J, Soárez PC, Novaes HM (2008) Rotavirus morbidity and mortality in children in Brazil. Rev Pan Am de Salud Publica 23:92–100
Flannery B, Samad S, de Moraes JC, Tate JE, Donovaro-Holliday MC, de Oliveira LH et al (2013) Uptake of oral rotavirus vaccine and timeliness of routine immunization in Brazil’s National Immunization Program. Vaccine 31:1523–1528
Linhares AC, Justino MCA (2014) Rotavirus vaccination in Brazil: effectiveness and health impact seven years post-introduction. Expert Rev Vaccines 13:43–57
Costa I, Linhares AC, Cunha MH, Tuboi S, Argüello DF, Justino MC et al (2016) Sustained decrease in gastroenteritis-related deaths and hospitalizations in children less than 5 years of age after the introduction of rotavirus vaccination: a time-trend analysis in Brazil (2001–2010). Pediatr Infect Dis J 35:e180–e190
do Carmo GM, Yen C, Cortes J, Siqueira AA, de Oliveira WK, Cortez-Escalante JJ et al (2011) Decline in diarrhea mortality and admissions after routine childhood rotavirus immunization in Brazil: a time-series analysis. PLoS Med 8:e1001024
Fernandes EG, Sato HK, Leshem E, Flannery B, Konstantyner TC, Veras MA et al (2014) Impact of rotavirus vaccination on diarrhea-related hospitalizations in São Paulo state, Brazil. Vaccine 32:3402–3408
Lanzieri TM, Linhares AC, Costa I, Kolhe DA, Cunha MH, Ortega-Barria E et al (2011) Impact of rotavirus vaccination on childhood deaths from diarrhea in Brazil. Int J Infect Dis 15:e206–e210
Sáfadi MA, Berezin EN, Munford V, Almeida FJ, de Moraes JC, Pinheiro CF et al (2010) Hospital-based surveillance to evaluate the impact of rotavirus vaccination in São Paulo, Brazil. Pediatr Infect Dis J 29:1019–1022
Estes MK, Greenberg HB (2013) Rotaviruses. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA et al (eds) Fields virology, 6th edn. Wolkers Kluwer Health, Philadelphia, pp 1347–1401
Jayaram H, Estes MK, Prasad BV (2004) Emerging themes in rotavirus cell entry, genome organization, transcription and replication. Virus Res 101:67–81
Rotavirus Classification Working Group: RCWG. [Online]. https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg. Accessed 24 July 2018
Bányai K, László B, Duque J, Steele AD, Nelson EA, Gentsch JR et al (2012) Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: insights for understanding the impact of rotavirus vaccination programs. Vaccine 30(Suppl 1):A122–A130
Gentsch JR, Laird AR, Bielfelt B, Griffin DD, Bányai K, Ramachandran M et al (2005) Serotype diversity and reassortment between human and animal rotavirus strains: implications for rotavirus vaccine programs. J Infect Dis 192(Suppl 1):S146–S159
Taniguchi K, Urasawa T, Kobayashi N, Gorziglia M, Urasawa S (1990) Nucleotide sequence of VP4 and VP7 genes of human rotaviruses with subgroup I specificity and long RNA pattern: implication for new serotype specificity. J Virol 64:5640–5644
Gómez MM, Resque HR, Volotão EM, Rose TL, Marques da Silva MF, Heylen E, Zeller M, Matthijnssens J, Leite JPG (2014) Distinct evolutionary origins of G12P[8] and G12P[9] group A rotavirus strains circulating in Brazil. Infect Genet Evol 28:385–388
Rahman M, Sultana R, Ahmed G, Nahar S, Hassan ZM, Saiada F et al (2007) Prevalence of G2P[4] and G12P[6] rotavirus, Bangladesh. Emerg Infect Dis 13:18–24
De Grazia S, Giammanco GM, Potgieter CA, Mthhhijnssens J, Banyai K, Platia MA et al (2010) Unusual assortment of segments in 2 rare human rotavirus genomes. Emerg Infect Dis 16:859–862
Martella V, Ciarlet M, Camarda A, Pratelli A, Tempesta M, Greco G et al (2003) Molecular characterization of the VP4, VP6, VP7, and NSP4 genes of lapinde rotaviruses identified in Italy: emergence of a novel VP4 genotype. Virology 314:358–370
Mochizuki M, Nakagomi T, Nakagomi O (1997) Isolation from diarrheal and asymptomatic kittens of three rotavirus strains that belong to the AU-1 genogroup of human rotaviruses. J Clin Microbiol 35:1272–1275
Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Bányai K, Estes MK et al (2008) Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol 153:1621–1629
Trojnar E, Sachsenröder J, Twardziok S, Reetz J, Otto PH, Johne R (2013) Identification of an Avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J Gen Virol 94:136–142
Gurgel RQ, Cuevas LE, Vieira SC, Barris VC, Fontes PB, Salustino EF et al (2007) Predominance of rotavirus P[4]G2 in a vaccinated population, Brazil. Emerg Infect Dis 13:1571–1573
Nakagomi T, Cuevas LE, Gurgel RG, Elrokhsi SH, Belkhir YA, Abugalia M et al (2008) Apparent extinction of non-G2 rotavirus strains from circulation in Recife, Brazil, after the introduction of rotavirus vaccine. Arch Virol 153:591–593
Carvalho-Costa FA, de Mello Volotão E, de Assis RM, Fialho AM, de Andrade JDS, Rocha LN et al (2011) Laboratory-based rotavirus surveillance during the introduction of a vaccination program, Brazil. Pediatr Infect Dis J 30(1):35–41
Guerra SF, Linhares AC, Mascarenhas JD, Oliveira A, Justino MC, Soares LS et al (2015) Rotavirus strain surveillance for three years following the introduction of rotavirus vaccine into Belém, Brazil. J Med Virol 87:1303–1310
Luchs A, Audrey C, Morillo SG, Carmona RCC, Timenetsky MCST (2015) Rotavirus genotypes circulating in Brazil, 2007–2012: implications for the vaccine program. Rev Inst Med Trop São Paulo 57:305–313
Neves MA, Pinheiro HH, Silva RS, Linhares AC, Silva LD, Gabbay YB et al (2016) High prevalence of G12P[8] rotavirus strains in Rio Branco, Acre, Western Amazon, in the post-rotavirus vaccine introduction period. J Med Virol 88:782–789
Justino MC, Linhares AC, Lanzieri TM, Miranda Y, Mascarenhas JD, Abreu E et al (2011) Effectiveness of the monovalent G1P[8] human rotavirus vaccine against hospitalization for severe G2P[4] rotavirus gastroenteritis in Belém, Brazil. Pediatr Infect Dis J 30:396–401
Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, na der Noordaa J (1990) Rapid and simple method for purification of nucleic acids. J Clin Microbiol 28:495–503
Pereira HG, Azeredo RS, Leite JP, Candeias JA, Rácz ML, Linhares AC et al (1983) Electrophoretic study of the genome of human rotaviruses from Rio de Janeiro, São Paulo, and Pará, Brazil. J Hyg (Lond) 90:117–125
Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J et al (1992) Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol 30:1365–1373
Gouvea V, Glass RI, Woods P, Taniguchi K, Clark HF, Forrester B et al (1990) Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol 28:276–282
Gouvea V, Santos N, Timenetsky MC (1994) VP4 typing of bovine and porcine group A rotaviruses by PCR. J Clin Microbiol 32(5):1333–1337
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinform Oxf Engl 28:1647–1649
Atschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Both GW, Siegman LJ, Bellamy AR, Ikegami N, Shatkin AJ, Furuichi Y (1984) Comparative sequence analysis of rotavirus genomic segment 6 the gene specifying viral subgroups 1 and 2. J Virol 1984(51):97–101
Cunliffe NA, Das BK, Ramachandran M, Bhan MK, Glass RI, Gentsch JR (1997) Sequence analysis demonstrates that VP6, NSP1 and NSP4 genes of Indian neonatal rotavirus strain 116E are of human origin. Virus Genes 15:39–44
Matthijnssens J, Rahman M, Martella V, Xuelei Y, De Vos S, De Leener K et al (2006) Full genomic analysis of human rotavirus strain B4106 and lapine rotavirus strain 30/96 provides evidence for interspecies transmission. J Virol 80:3801–3810
Varghese V, Das S, Singh NB, Kojima K, Bhattacharya SK, Krishnan T et al (2004) Molecular characterization of a human rotavirus reveals porcine characteristics in most of the genes including VP6 and NSP4. Arch Virol 149:155–172
da Soares LS, dos Lobo PS, Mascarenhas JDP, Neri DL, dos Guerra SFS, de Oliveira AD et al (2012) Identification of lineage III of G12 rotavirus strains in diarrheic children in the Northern region of Brazil between 2008 and 2010. Arch Virol 157:135–139
Matthijnssens J, Heylen E, Zeller M, Rahman M, Lemey P, Van Ranst M (2010) Phylodynamic analyses of rotavirus genotypes G9 and G12 underscore their potential for swift global spread. Mol Biol Evol 27:2431–2436
Soares LS, Guerra SFS, de Oliveira ASL, dos Santos FS, Menezes EMFC, Mascarenhas JDP et al (2014) Diversity of rotavirus strains circulating in Northern Brazil after introduction of a rotavirus vaccine: high prevalence of G3P[6]. J Med Virol 86:1065–1072
da Silva MF, Fumian TM, de Assis RM, Fialho AM, Carvalho-Costa FA, da Silva Ribeiro de Andrade J et al (2017) VP7 and VP8* genetic characterization of group A rotavirus genotype G12P[8]: emergence and spreading in the Eastern Brazilian coast in 2014. J Med Virol 89:64–70
Dia ML, Diop A, Sonko MA, Bâ M, Cissá MF (2015) First report of gastroenteritis by genotype G12 rotavirus in Dakar, Senegal. New Microbes New Infect 20:30–32
Kang G, Desai R, Arora R, Chitambar S, Naik TN, Krishnan T et al (2013) Diversity of circulating rotavirus strains in children hospitalized with diarrhea in India, 2005–2009. Vaccine 31:2879–2883
Nakagomi T, Do LP, Agbemabiese CA, Kaneko M, Gauchan P, Doan YH et al (2016) Whole-genome characterisation of G12P[6] strains possessing two distinct genotype constellations co-circulating in Blantyre, Malawi, 2008. Arch Virol 162:213–226
Pun SB, Nakagomi T, Sherchand JB, Pandey BD, Cuevas LE, Cunliffe NA et al (2007) Detection of G12 human rotaviruses in Nepal. Emerg Infect Dis 13:482–484
De Vos B, Han HH, Bouckenooghe A, Debrus S, Gillard P, Ward R et al (2009) Live attenuated human rotavirus vaccine, RIX4414, provides clinical protection in infants against rotavirus strains with and without shared G and P genotypes: integrated analysis of randomized controlled trials. Pediatr Infect Dis J 28:261–266
Patel M, Pedreira C, De Oliveira LH, Tate J, Leshem E, Mercado J et al (2016) Effectiveness of pentavalent rotavirus vaccine against a diverse range of circulating strains in Nicaragua. Clin Infect Dis 62(Suppl 2):S127–S132
Steele AD, Neuzil KM, Cunliffe NA, Madhi SA, Bos P, Ngwira B et al (2012) Human rotavirus vaccine RotarixTM provides protection against diverse circulating rotavirus strains in African infants: a randomized controlled trial. BMC Infect Dis 12:2013. https://doi.org/10.1186/1471-2334-12-213
Castello AA, Nakagomi O, Jiang B, Kang JO, Glass RI, Glikman G et al (2009) Characterization of genotype P[9]G12 rotavirus strains from Argentina: high similarity with Japanese and Korean G12 strains. J Med Virol 81:371–381
Pietruchinski E, Benati F, Lauretti F, Kisielius J, Ueda M, Volotão EM et al (2006) Rotavirus diarrhea in children and adults in a southern city of Brazil in 2003: distribution of G/P types and finding of a rare G12 strain. J Med Virol 78:1241–1249
Pongsuwana Y, Guntapong R, Chiwakul M, Tacharoenmuang R, Onvimala N, Wakuda M et al (2002) Detection of a human rotavirus with G12 and P[9] specificity in Thailand. J Clin Microbiol 40:1390–1394
Shinozaki K, Okada M, Nagahima S, Kaiho I, Taniguchi K (2004) Chracterization of human rotavirus strains with G12 and P[9] detected in Japan. J Med Virol 73:612–616
Santos N, Volotão EM, Soares CC, Campos GS, Sardi SI, Hoshino Y (2005) Predominance of rotavirus genotype G9 during the 1999, 2000, and 2002 seasons among hospitalized children in the city of Salvador, Bahia, Brazil: implications for future vaccine strategies. J Clin Microbiol 43:4064–4069
Khamrin P, Niwat M, Peerakome S, Tonusin S, Phan TG, Okitsu S et al (2007) Molecular characterization of rare G3P[9] rotavirus strains isolated from children hospitalized with acute gastroenteritis. J Med Virol 79:843–851
Piekarska A, Kacerka A, Majda-Stanislawska E, Jówiak B, Sidorkiewicz M (2015) Predominance of genotype P[9]G3 in rotavirus gastroenteritis in Polish children. Arch Med Sci 11:577–583
Tsugawa T, Rainwater-Lovett K, Tsutsumi H (2015) Human G3P[9] rotavirus strains possessing an identical genotype constellation to AU-1 isolated at high prevalence in Brazil, 1997–1999. J Gen Virol 96:590–600
Martella V, Potgieter AC, Lorusso E, Grazia S, Giammanco GM, Matthijnssens J et al (2011) A feline rotavirus G3P[9] carries traces of multiple reassortment events and resemble rare human G3P[9] rotaviruses. J Gene Virol 92:1214–1221
Nakagomi O, Nakagomi T, Hoshino Y, Flores J, Kapikian AZ (1987) Genetic analysis of a human rotavirus that belongs to subgroup I but has an RNA pattern typical of subgroup II human rotaviruses. J Clin Microbiol 25:1159–1164
Nakagomi T, Nakagomi O (1989) RNA–RNA hybridization identifies a human rotavirus that is genetically related to a feline rotavirus. J Virol 63:1431–1434
Bezerra DA, Guerra SF, Serra AC, Fecury PC, Bandeiras RS, Penha ET et al (2017) Analysis of a genotype G3P[9] rotavirus strain that shows evidence of multiple reassortment events between animal and human rotaviruses. J Med Virol 89:974–981
Espínola EE, Parra GI, Russomando G, Arbiza J (2008) Genetic diversity of the VP4 and VP7 genes affects the genotyping of rotaviruses: analysis of Paraguayan strains. Infect Genet Evol 8:94–99
Solberg OD, Hasing ME, Trueba G, Eisenberg JN (2009) Characterization of novel VP7, VP4, and VP6 genotypes of a previously untypeable group A rotavirus. Virology 385:58–67
Iturriza-Gómara M, Isherwood B, Desselberger U, Gray J (2001) Reassortment in vivo: driving force for diversity of human rotavirus strains isolated in the United Kingdom between 1995 and 1999. J Virol 75(8):3696–3705
Arana A, Montes M, Jere KC, Alkorta M, Itrriza-Gómara M, Cilla G (2015) Emergence and spread of G3P[8] rotaviruses possessing an equine-like VP7 and a DS-1-like genetic backbone in the Basque country (North of Spain). Infect Genet Evol 44:137–140
Cowley D, Donato CM, Roczo-Farkas S, Kirkwood CD (2016) Emergence of a novel equine-like G3P[8] inter-genogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J Gen Virol 97:403–410
Guerra SF, Soares LS, Lobo OS, Penha Júnior ET, Souza Júnior EC, Bezerra DA et al (2016) Detection of a novel equine-like G3 rotavirus associated with acute gastroenteritis in Brazil. J Gen Virol 97:3131–3138
Luchs A, da Costa AC, Cilli A, Komninakis SCV, Carmona RCC, Boen L et al (2018) Spread of the emerging equine-like G3P[8] DS-1-like genetic backbone rotavirus strain in Brazil and identification of potential genetic variants. J Gen Virol. https://doi.org/10.1099/jgv.0.001171
Perkins C, Mijatovic-Rustempasic S, Ward ML, Cortese MM, Bowen MD (2017) Genomic characterization of the first equine-like G3P[8] rotavirus strain detected in the United States. Genome Announc 5(47):e01341-17
Roczo-Farkas S, Kirkwood CD, Bines JE, Australian Rotavirus Surveillance Group (2015) Australian Rotavirus Surveillance Program annual report. Commun Dis Intell Q Rep 40:E527–E538
Komoto S, Tacharoenmuang R, Guntapong R, Ide T, Tsuji T, Yoshikawa T et al (2016) Reassortment of human and animal rotavirus gene segments in emerging DS-1-like G1P[8] rotavirus strains. PLoS One 11:e0148416
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Reimar Johne.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guerra, S.F.S., Fecury, P.C.M.S., Bezerra, D.A.M. et al. Emergence of G12P[6] rotavirus strains among hospitalised children with acute gastroenteritis in Belém, Northern Brazil, following introduction of a rotavirus vaccine. Arch Virol 164, 2107–2117 (2019). https://doi.org/10.1007/s00705-019-04295-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04295-w