
Abstract
A novel ssRNA (+) virus with molecular properties typical of members of the genus Vitivirus (family Betaflexiviridae; subfamily Trivirinae) was discovered by high-throughput sequencing in samples of the American hybrid bunch grape cultivar Blanc du Bois in Texas. The results were independently confirmed by Sanger sequencing of the virus isolate, whose genome length is 7,387 nt, excluding the polyA tail. The genome sequence contains five ORFs that are homologous and phylogenetically related to ORFs of grapevine-infecting vitiviruses. The name “grapevine virus M” is proposed for this new virus, whose sequence divergence exceeds the current ICTV species demarcation threshold for the genus Vitivirus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The genus Vitivirus (family Betaflexiviridae; subfamily Trivirinae) consists of several ssRNA (+) viruses characterized from grapevine and other perennial plants. To date, 15 species have been assigned to the genus (https://talk.ictvonline.org/files/master-species-lists/m/msl/7992). Eleven vitiviruses were discovered in grapevine (Vitis spp.), the host that inspired the genus epithet. The grapevine-infecting vitiviruses may not elicit discernible foliar symptoms, but infection by some, such as grapevine virus A (GVA) and grapevine virus B (GVB), has been associated with detrimental modifications of the vine woody cylinder and Shiraz disease [1]. Mealybugs (Hemiptera: Pseudococcidae) and/or soft scale insects (Hemiptera: Coccidae) are vectors for GVA, GVB and grapevine virus E (GVE) [1] whereas aphid transmission has been reported for heracleum latent virus (HLV) and mint virus 2 (MV-2) [2, 3]. The genomes of vitiviruses are organized into five ORFs, except for those of arracacha virus V (AVV) and HLV, which lack an ORF2. The ORF1 encodes a large replication-associated protein (REP), ORF2 encodes a hypothetical protein of unknown function, ORF3 encodes a movement protein (MP), ORF4 encodes the coat protein (CP), and ORF5 encodes a nucleic-acid-binding protein (NABP) [4].
In May 2018, we explored the virome of a 32-year-old Blanc du Bois (Vitis spp.: ‘Florida D 6-148’ × ‘Cardinal’) vineyard located in Austin County, Texas. Leaf tissue samples (with intact petioles) were collected from 51 vines across the vineyard, flash frozen in liquid N2, and stored in -80 °C until processing. Total RNA was extracted from each of the 51 samples using a SpectrumTM Plant Total RNA Kit (Sigma-Aldrich) and equimolar amounts of RNA from each sample were pooled into a single RNA sample, which was subjected to high-throughput sequencing (HTS) on an Illumina NextSeq 500 platform [5]. The approximately 23.5 million raw HTS reads generated from the sample (TX-WAT) were bioinformatically processed as described earlier [6]. The analysis revealed contigs specific to five viruses and three viroids, among which only 10 contigs (ranging from 206 to 4,405 nt) showed similarities to vitivirus sequences, particularly the recently characterized grapevine virus H (GVH) [7] (query coverage, 100%; average identity, 68%). A quick screen of each of the 51 RNA samples by RT-PCR with two primer pairs designed from the HTS-derived sequences (data not shown) revealed a 11.8% (6/51) incidence of the vitivirus.
Independently, the near-complete genome of the virus was amplified from three positive samples as two pieces of overlapping DNA fragments using two sets of primer pairs designed from the HTS-derived sequences of TX-WAT. The primer pair GVM-264v (5’-CCAgTTAgCAgCgAAAggTAT) and GVM-4086c (5’-gTTgTACCTCCTTCggTTCTTC) amplified a 3,823-bp DNA fragment specific to the REP, and the primer pair GVM-3820v (5’-CCgACgAAgACCCATTTgTA) and GVM-7211c (5’-CTCTACCgTATTTCACCCAgTC) amplified a 3,392-bp DNA fragment that encompassed the C-terminal sequences of the REP and other genes. RT-PCR, cloning, and Sanger sequencing were done as described earlier [8]. Sequences of the 5’ and 3’ extremities of the virus were determined using a Clontech SMARTer RACE 5’/3’ kit (Takara-Bio). Pairwise comparisons of the fragment-specific sequences derived from the three samples showed > 99% sequence identity among them; hence, the genome sequence of the virus was assembled using BioEdit software [9] from fragments obtained from one of the isolates. The programs ORF Finder and SMART BLAST (https://www.ncbi.nlm.nih.gov/orffinder/) were used to predict and verify potential protein-encoding segments of the DNA sequence.
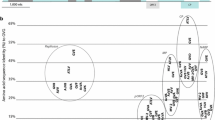
The complete genome of the vitivirus isolate TX-WAT, excluding the polyA tail, was determined to be 7,387 nt in length (MK492703), a typical size for a vitivirus genome [4]. The sequence obtained by Sanger sequencing shared 99.7% nt identity with the HTS-derived sequence (~ 88% genome coverage), thus validating the HTS results. The genome organization and lengths of the encoded genes (Fig. 1) are typical for members of the genus Vitivirus [1, 4]. The REP-encoding ORF1 is 5,151 nt long (1,716 amino acids [aa]; 194.5 kDa), and it shared the highest nt (65.8%) and aa (74.7%) sequence identity with the REP of GVH; it contains all of the typical conserved motifs for members of the family Betaflexiviridae. ORF2 is 492 nt long (163 aa; 18.1 kDa) and encodes a hypothetical protein of unknown function that shares 55% nt and 65.6% aa sequence identity with ORF2 of GVH. ORF3 encodes an 804-nt MP (267 aa; 29.7 kDa) that shares 70.2% nt and 69.7% aa sequence identity with the MP of GVH. The CP-encoding ORF4 is 594 nt long (197 aa; 21.5 kDa) and it shared greatest similarity (nt: 78.7%; aa: 88.8%) with the CP of GVH. ORF5 encodes a 336-nt NABP (111 aa; 13.0 kDa) that shares 71.9% nt and 70.5% aa sequence identity with the NABP of GVH. Phylogenetic analysis of the predicted REP and CP aa sequences supports the placement of TX-WAT within the genus Vitivirus in the same clade as GVH and other grape-infecting vitiviruses (Fig. 2).

Genome organization of grapevine virus M. The positions of the five open reading frames are shown as rectangular boxes. REP, replicase; ORF2, a hypothetical protein of unknown function; MP, movement protein; CP, coat protein; NABP, nucleic-acid-binding protein; A(n), polyA 3’ tail

Neighbor-joining phylogenetic trees (1,000 bootstrap replications) based on the amino acid sequences of the replicase (A) and coat protein (B) of members of the genus Vitivirus. The proteins of the newly characterized grapevine virus M (GVM) are indicated in bold font with a colored background
The taxonomic status of isolate TX-WAT was interrogated vis-à-vis the current ICTV species demarcation threshold for the family Betaflexiviridae [4]. While an analysis of the CP sequences of TX-WAT would suggest that it is a variant of GVH, the nt and aa sequence identity levels in its REP are well below the set threshold of < 72% nt or < 80% aa identity for new species [4]. A similar situation exists for the recently characterized GVL isolate KA (MH643739), which also had sequence identity values above (nt: 71.6%; aa: 77.8%) and below (nt: 64.2%; aa: 69%) the species demarcation threshold values when compared with GVE isolate TvAQ7 in the taxonomically relevant CP and REP, respectively. Thus, we posit that isolate TX-WAT represents a new species in the genus Vitivirus, and we propose the name “grapevine virus M” (GVM) for this new virus. Further evidence that GVM is distinct from GVH could be seen in the relatively low levels of nt and aa sequence similarity between their ORF2, MP and NABP and the shorter length (12 nt) of its RACE-verified 3’ UTR sequence.
In future work, we will explore the biological properties of GVM and attempt to develop robust diagnostic assays for its accurate discrimination from GVH and other grapevine-infecting vitiviruses in epidemiological studies.
References
Minafra A, Mawassi M, Goszczynski D, Saldarelli P (2017) Grapevine vitiviruses. In: Meng B, Martelli G, Golino D, Fuchs M (eds) Grapevine viruses: molecular biology, diagnostics and management. Springer, Cham, pp 229–256
Tzanetakis IE, Postman JD, Martin RR (2007) Identification, detection and transmission of a new vitivirus from Mentha. Arch Virol 152:2027–2033
Bem F, Murant AF (1979) Transmission and differentiation of six viruses infecting hogweed (Heracleum sphondylium) in Scotland. Ann Appl Biol 92:237–242
Adams MJ, Candresse T, Hammond J, Kreuze JF, Martelli GP, Namba S, Pearson MN, Ryu KH, Saldarelli P, Yoshikawa N (2012) Family betaflexiviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy, vol 152. 9th Report of the ICTV. Elsevier, Academic Press, Amsterdam, pp 2027–2033
Al Rwahnih M, Alabi OJ, Westrick NM, Golino D, Rowhani A (2017) Description of a novel monopartite geminivirus and its defective subviral genome in grapevine. Phytopathology 107:240–251
Al Rwahnih M, Rowhani A, Westrick N, Stevens K, Diaz-Lara A, Trouillas F, Preece J, Kallsen CE, Farrar K, Golino DA (2018) Discovery of viruses and virus-like pathogens in pistachio using high-throughput sequencing. Plant Dis 102:1419–1425
Candresse T, Theil S, Faure C, Marais A (2017) Determination of the complete genomic sequence of grapevine virus H, a novel vitivirus infecting grapevine. Arch Virol 163:277–280
Yahaya A, Dangora DB, Kumar PL, Alegbejo MD, Gregg L, Alabi OJ (2019) Prevalence and genome characterization of field isolates of sugarcane mosaic virus (SCMV) in Nigeria. Plant Dis. https://doi.org/10.1094/PDIS-08-18-1445-RE (Accepted for publication)
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Acknowledgements
The authors acknowledge funding support from the Texas Wine Grape Growers Association through Senate Bill 881 (SB881). We thank an anonymous grower for his cooperation and access to the Blanc Du Bois vineyard. We thank Mr. Jonathan Motloch, Ms. Regina Hernanadez and Ms. Cecilia Villegas (Texas A&M University System) for help with sample processing, and Dr. Minsook Hwang and Dr. Kristian Stevens (UC Davis Foundation Plant Services) for help with HTS library preparation and bioinformatics analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no conflict of interest. The funders had no role in the design of the study, collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Ioannis E. Tzanetakis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alabi, O.J., McBride, S., Appel, D.N. et al. Grapevine virus M, a novel vitivirus discovered in the American hybrid bunch grape cultivar Blanc du Bois in Texas. Arch Virol 164, 1739–1741 (2019). https://doi.org/10.1007/s00705-019-04252-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04252-7