
Abstract
In this study, we isolated a novel virulent Escherichia phage, SRT7. Its genome is a double-stranded linear DNA molecule containing 39,883 bp. Direct terminal repeats with a length of 175 bp, are present at both ends of the genome. The G+C content is 50.54%. Forty-seven putative protein coding genes were identified. No tRNA or rRNA genes were identified. Comparative genomic analysis revealed that phage SRT7 is a novel member of the T7-like phage cluster, but it forms a singleton subcluster.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bacteriophages, discovered by Frederick W. Twort in 1915 and Félix d’Hérelle in 1917, are a group of viruses that infect bacteria. They are the most numerous organisms on Earth, and there are estimated to be about 1031 bacteriophages in the world [1]. Some scientists consider phages to be the dark matter of the biosphere [2]. Our understanding about the origin, evolution, and relationships of different phages is still limited. Comparative genomic analysis of novel phages is considered one of the key approaches to investigating the origin, evolution, and diversity of phages [2]. As a model microorganism, Escherichia coli is one of the most studied bacteria in molecular biology and microbiology. Furthermore, it only needs to acquire certain genetic elements to become a pathogen that it can cause diseases that affect animals and humans [3]. Compared to what is known about E. coli itself, there is relatively little information on Escherichia phages. One of the best characterized Escherichia phages is T7. T7-like phages are viruses that infect E. coli and some other enteric bacteria. The evolutionary process of T7-like phages is still a mystery. Isolation, sequencing, and genomic analysis of novel T7-like phages can help not only in understanding their evolution but also in improving Escherichia phage therapy.
In this study, using E. coli BL21 as the host organism, Escherichia phage SRT7 was isolated from a soil sample collected from a refuse dump (36°62′N, 116°96′E) of the University of Jinan, China. Using a standard protocol [4], phage particles were purified from bacterial lysates with 2.5 M NaCl and 20% PEG 8000. Using a previously described method [5], genomic DNA was extracted from the purified phage particles. The whole genome was sequenced at the Realbio Genomics Institute (Shanghai, China), using the Illumina HiSeq PE150 platform. Complete genome sequences were assembled using CLC genomics workbench 11 (Aarhus, Denmark). Direct terminal repeats were found at both ends of phage SRT7 genome. Both repeats are 175 bp in length. The genome of SRT7 is a 39,883-bp linear dsDNA molecule. Coding sequences were identified using Glimmer v3.02 [6]. The functions of the encoded proteins were predicted by screening against the Non-Redundant Protein Database [7] using blastp [8]. The presence of tRNA and rRNA genes was detected by tRNAscan-SE [9] and RNAmmer [10], respectively. The NCBI Blastn tool was used for comparative genomic analysis of SRT7 and T7-like phages, and this was followed by clicking on the “Distance tree of results” link. A genome-wide phylogenetic tree of 171 T7-like phages and SRT7 was constructed using the neighbor-joining method [11]. A sequence comparison of the SRT7 with T7 genomes was made using Easyfig v2.2.3 [12].
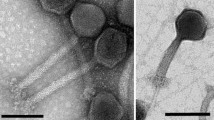
Escherichia phage SRT7 formed clear plaques with a diameter of about 1 cm. The results indicated that phage SRT7 is a virulent phage. Its genome is a double-stranded linear DNA molecule containing 39,883 bp. Direct terminal repeats of 175 bp are present at both ends of genome. The G+C content is 50.54%. No tRNA and rRNA genes were identified. The genome contains 47 putative protein coding genes. Forty of these proteins showed sequence similarity to other phage proteins, 27 of which could be assigned a function based on blastp (Table S1).
The functional proteins were classified into three modules, namely, a host lysis module, a phage structure and packaging module, and a DNA metabolism module (Fig. 1). The host lysis module was restricted to gp2, gp4 and gp29, whose protein products had sequence similarity to endopeptidase of Escherichia phage T3, lysis protein of Escherichia phage T7, and lysozyme of Erwinia phage vB_EamP-L1, respectively. The phage structure and packaging module had thirteen genes, gp1, gp3, gp5, gp6, gp7, gp8, gp9, gp10, gp11, gp13, gp14, gp15, and gp16, which encode DNA maturation protein, DNA packaging protein A, tail fiber protein, internal virion protein D, internal virion protein C, internal virion protein B, internal virion protein A, tail tubular protein B, tail tubular protein A, major capsid protein, capsid assembly protein, head-to-tail joining protein, and host range protein, respectively. The DNA metabolism module consists of nine genes, namely, gp20, gp24, gp28, gp31, gp32, gp33, gp36, gp40, and gp41, which code for exodeoxyribonuclease, DNA polymerase, primase/helicase protein, endonuclease I, single-stranded DNA-binding protein, bacterial RNA polymerase inhibitor, ATP-dependent DNA ligase, and DNA-directed RNA polymerase, respectively. In addition, a protein kinase (encoded by gp42) with sequence similarity (44% identity) to protein kinase Gp 0.7 of Escherichia phage T7 was identified. As a host shutoff protein, the protein kinase can target various components of translation and transcription [13, 14] and plays an important role in the process of phage-host interaction. Gp47 has been annotated as an S-adenosyl-L-methionine hydrolase.

Functional gene map and comparative genomic analysis of Escherichia phages SRT7 and T7. The red arrows represents the host lysis module; blue arrows represent the phage structure and packaging module; green arrows represent the DNA metabolism module; black arrows represent genes that cannot be classified into the three functional modules; gray arrows represent genes without similarity to sequences in the Non-Redundant Protein Database. Conserved genes of phage T7 are arranged in three gene clusters (class I, class II, and class III). Red shading indicates that the DNA fragments are highly homologous
Many other T7-like phages have been isolated previously [15]. In a NCBI ‘blastn’ comparison, the genome sequences of 171 phages, including Yersinia phage vB_Ye nP_AP10, which is the most closely related to SRT7 (coverage, 53%; identity, 71%), and Escherichia phage T7 (coverage, 47%; identity, 74%), showed nucleotide sequence similarity to SRT7, indicating that phage SRT7 belongs to the T7-like phage cluster. The species demarcation criterion for phage is set at 95% identity at the nucleotide level [16], indicating that Escherichia phage SRT7 is a novel phage. Comparative genomic analysis also shows that there are differences in the gene arrangement of SRT7 and T7 [17] (Fig. 1). In 2014, Julianne H. Grose and Sherwood R. Casjens carried out a genomic comparison of tailed phages that infect Enterobacteriaceae hosts [18]. The T7-like phage cluster was divided into twelve subclusters (phages of eight subclusters, A, B, C, D, E, F, G and H, can infect Enterobacteriaceae hosts and four subclusters that infect hosts outside the family Enterobacteriaceae). The genome-wide phylogenetic tree of 171 phages and SRT7 showed that SRT7 does not belong to any of the existing T7-like subclusters (Fig. 2). The results show that SRT7 is a novel phage and forms a novel singleton subcluster of the T7-like cluster.

Phylogenetic tree of SRT7 and 171 phages that have nucleotide sequence similarity to the SRT7 genome. T7-like I-subcluster phages can infect Vibrio; T7-like J-subcluster phages can infect Pseudomonas; T7-like K-subcluster phages can infect Ralstonia; phages of subclusters A, B, C, D, E, F, G and H can infect Enterobacteriaceae hosts
The GenBank accession number for Escherichia phage SRT7 is MH370477.
References
Hendrix RW, Smith MCM, Burns RN, Ford ME, Hatfull GF (1999) Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natil Acad Sci 96(5):2192–2197
Hatfull GF (2015) Dark matter of the biosphere: the amazing world of bacteriophage diversity. J Virol 89(16):8107–8110
Croxen MA, Finlay BB (2010) Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 8(1):26–38
Russell DW, Sambrook J (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Fan X, Yan J, Xie L, Zeng L, Young RF, Xie J (2015) Genomic and proteomic features of mycobacteriophage SWU1 isolated from China soil. Gene 561(1):45–53
Delcher AL, Bratke KA, Powers EC, Salzberg SL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23(6):673–679
Pruitt KD, Tatusova T, Maglott (2006) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35(suppl_1):D61–D65
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36(suppl_2):W5–W9
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33(Web Server Issue):W686–W689
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9):3100–3108
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Sullivan MJ, Petty NK, Beatson SA (2011) Easyfig: a genome comparison visualizer. Bioinformatics 27(7):1009–1010
Severinova E, Severinov K (2006) Localization of the Escherichia coli RNA polymerase beta’ subunit residue phosphorylated by bacteriophage T7 kinase Gp0.7. J Bacteriol 188(10):3470–3476
Robertson ES, Aggison LA, Nicholson AW (1994) Phosphorylation of elongation factor G and ribosomal protein S6 in bacteriophage T7-infected Escherichia coli. Mol Microbiol 11(6):1045–1057
Kabanova A, Shneider M, Bugaeva E, Ha VTN, Miroshnikov K, Korzhenkov A, Kulikov E, Toschakov S, Ignatov A, Miroshnikov K (2018) Genomic characteristics of vB_PpaP_PP74, a T7-like Autographivirinae bacteriophage infecting a potato pathogen of the newly proposed species Pectobacterium parmentieri. Arch Virol 163(6):1691–1694
Adriaenssens E, Brister JR (2017) How to name and classify your phage: an informal guide. Viruses 9(4):70
Dunn JJ, Studier FW, Gottesman M (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol 166(4):477–535
Grose JH, Casjens SR (2014) Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468–470:421–443
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was funded by the National Natural Science Foundation (31600148, 31100088, 31600066), the Foundation of the University of Jinan (XBS1519, XKY1633), the Shandong Excellent Young Scientist Award Fund (BS2014YY031), and the Shandong Provincial Natural Science Foundation (ZR2016CB20).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Chan-Shing Lin.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, K., Song, S., Zhao, Z. et al. The complete genome sequence of Escherichia phage SRT7, a novel T7-like phage. Arch Virol 164, 1217–1219 (2019). https://doi.org/10.1007/s00705-019-04182-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04182-4