
Abstract
Human bocavirus (HBoV) is a recently discovered parvovirus associated with respiratory and gastroenteric infections in children. To date, four distinct subtypes have been identified worldwide. HBoV1 is the most frequently detected bocavirus in clinical samples derived from the respiratory tract. HBoV has a single-stranded DNA genome, which encodes two nonstructural proteins, NS1 and NP1, and two structural proteins, VP1 and VP2. Despite a large number of available HBoV sequences, the molecular evolution of this virus remains enigmatic. Here, we applied bioinformatic methods to measure the codon usage bias in 156 HBoV genomes and analyzed the factors responsible for preferential use of various synonymous codons. The effective number of codons (ENC) indicates a highly conserved, gene-specific codon usage bias in the HBoV genome. The structural genes exhibit a higher degree of codon usage bias than the non-structural genes. Natural selection emerged as dominant factor influencing the codon usage bias in the HBoV genome. Other factors that influence the codon usage include mutational pressure, gene length, protein properties, and the relative abundance of dinucleotides. The results presented in this study provide important insight into the molecular evolution of HBoV and may serve as a primer for HBoV gene expression studies and development of safe and effective vaccines to prevent infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human bocavirus (HBoV) was first identified in 2005 in the nasal washings of children suffering from idiopathic respiratory disease [2]. To date, four distinct subtypes of HBoV have been identified and classified as HBoV1-4 [3, 32]. HBoV1 is the most frequently detected bocavirus in human respiratory samples, accounting for 65-95% of all HBoV infections [61]. HBoV2–4 are mostly found in fecal samples and have been implicated in gastrointestinal infections [13, 26]. HBoV is a member of the family Parvoviridae, subfamily Parvovirinae, and genus Bocaparvovirus [50]. Phylogenetic analysis has indicated a close evolutionary relationship between HBoV and bocaviruses found in great apes [55]. Based on the viral NS1 protein sequence, HBoV1 and 3 as well as gorilla and chimpanzee bocaviruses have been classified as members of the primate bocaparvovirus 1 group, whereas HBoV2 and 4 have been classified as members of the primate bocaparvovirus 2 group [15].
Human bocavirus (HBoV) has a single-stranded DNA genome of approximately ~ 5.3 kb. The genome contains three open reading frames (ORF), encoding two structural proteins, VP1 and VP2, and two nonstructural proteins, NS1 and NP1. The coding sequence is flanked by inverted repeats that form hairpin-like structures and are essential for viral genome replication [33, 42]. The NS1 is a multifunctional protein that is required for viral DNA replication, gene expression, and immune evasion [38, 64]. The NP1 is a phosphoprotein and plays a role in nuclear localization, blocks interferon production by interacting with interferon regulatory factor 3 (IRF3), and induces both cell arrest and apoptosis in HeLa cells [39, 63, 69]. The capsid proteins VP1 and VP2 share a C-terminal region and differ only in the N-terminal region of VP1. The N-terminal portion of VP1 contains a unique phospholipase A motif that is essential for infectivity and facilitates the release of the virus from endocytic compartments to the nucleus of the host cell [24, 51]. VP2 upregulates the production of interferon beta (IFN-β) and represents the major antigenic determinant for serology and immunization [18, 41].
As obligate intracellular parasites, viruses rely exclusively on the translational machinery of the host cell for the synthesis of viral proteins. The efficiency of viral mRNA translation depends on factors such as the composition of amino acid/ tRNA pools and the codon frequency within the mRNA [1, 5, 9, 11, 17, 27, 28, 48]. Although the genetic code is essentially universal, the abundance of isoaccepting tRNAs varies between organisms, between the tissues of same organism, and between various stages of differentiation of same tissue [70]. These tRNA variations may explain, at least in part, spatial or temporal regulation of viral gene expression within host organisms [19, 60]. Viruses have evolved to take advantage of codon composition to regulate gene expression without having deleterious effects on host cells. In papillomaviruses, expression of the capsid proteins L1 and L2 is linked to the specific cell differentiation states in which particular aminoacyl-tRNAs are expressed [71, 74]. In hepatitis A virus, the presence of low-prevalence codons has been linked to slow protein translation and correct protein folding [14]. Human immunodeficiency virus and herpesviruses maintain codons in their genes that have a low prevalence in their host cells [7, 46].
Despite the availability of a large number of HBoV sequences in genome database, few data are available about the synonymous codon usage patterns of this virus. A study of nine Parvovirinae subfamily viruses revealed that the codon bias is greatly influenced by GC frequency [54]. The synonymous codon usage bias of 11 human bocaviruses suggested that mutational bias and gene function are the main factors that determine the codon usage bias in HBoV [72]. A more recent study on 58 Parvoviridae family viruses showed that natural selection rather than mutation pressure is the main factor that affects the codon bias [58]. HBoV is a serious public health risk and necessitate in-depth scientific studies to understand the evolution and pathogenic potential of this virus.
This study was undertaken to address following important issues related to HBoV evolution: (i) the extent of codon bias in the HBoV genome; (ii) inter and intra-genomic variations in codon usage bias; and (iii) forces that influence the evolution of codon bias. To address these questions, we have analyzed the nucleotide composition of 156 HBoV genomes, quantified the codon usage bias and performed a correlation analysis of various indices of codon usage bias to determine the extent and relative influence of different evolutionary pressures on HBoV codon usage. A better knowledge of codon usage bias is essential to understanding the processes governing HBoV evolution. Furthermore, such information is relevant to understanding the regulation of viral gene expression and also to vaccine design, where the efficient expression of viral proteins may be required to generate immunity [25]. Our results suggest a low but highly conserved gene-specific codon usage bias in the HBoV genome. Natural selection emerged as a key determinant of codon usage bias in the HBoV genome, whereas mutation pressure and gene function emerged as minor contributing factors that differentially affect the codon usage in different HBoV genes.
Materials and methods
Sequence data
The genome sequences in Fasta format were retrieved from the National Center for Biotechnology (NCBI) GenBank database (http://www.ncbi.nlm.nih.gov). Sequences were aligned using MEGA 7.0, and sequences with excessive gaps or incomplete ORFs were removed. Details of 156 HBoV isolates used in the study are provided in Table S1. To determine the genomic parameters and overall codon usage bias in the entire coding sequence (CDS), stop codons of NS1 and NP1 were removed and open reading frames (ORFs) were concatenated in the following order: NS1 + NP1 + VP1/2. Comparative analysis of nucleotide composition, codon usage bias, and forces shaping codon usage in different HBoV genes was carried out.
Nucleotide composition
The overall frequency of occurrence of mononucleotides (A, C, T, and G), the frequency of occurrence of mononucleotides at the third synonymous codon position (A3s, C3s, T3s and G3s), and the frequency of occurrence of G+C (GC1, GC2, GC12 and GC3s) (Table 1), and the observed frequency of dinucleotides (ApA, ApC, ApG, ApT, CpA, CpC, CpG, CpT, GpA, GpC, GpG, GpT, TpA, TpC, TpG and TpT) were calculated using default parameters in the SSE software package [59]. Expected dinucleotide frequencies were also calculated assuming random association of bases from the observed frequencies of each base. A ratio of observed to expected dinucleotide (O/E) frequencies was used for designation of over-representation or under-representation in terms of relative abundance compared with a random association of mononucleotides [36, 66].
Effective number of codons (ENC)
The ENC is a measure of the total number of different codons present in a sequence and shows the bias from an equal use of all synonymous codons for a given amino acid [47, 68]. The ENC values for HBoV CDS and individual genes were calculated using default parameters in the SSE software package [59].
Relative synonymous codon usage (RSCU)
The RSCU is one of the most widely used parameters for examining the overall synonymous codon usage variation among genes [56]. The RSCU is the number of times a codon appears in a gene divided by the number of expected occurrences under equal codon usage. Codons with RSCU values of < 1.0, 1.0, > 1.0 represent negative codon usage bias, no bias, and positive codon usage bias, respectively. The RSCU values of all HBoV sequences were calculated using the MEGA7.0 software package [35] (Table 2). Single codons ATG (Met) and TGG (Trp), and the termination codons TAA, TAG, and TGA are not expected to exhibit any usage bias and were therefore excluded from the analysis.
Codon adaptation index (CAI)
The CAI is a measure of the adaptiveness of a gene towards the codon usage of highly expressed genes [57]. The CAI values for all HBoV genes were calculated using human codon usage as a reference set in CAI calculator [49].
ENc-plot mapping analysis
An ENC-Plot (ENC vs. G3s) is commonly used to determine the effect of G+C compositional constraints on codon usage bias [68]. An ENC-plot is a comparison of the observed and expected distribution of genes base on GC3s on a single plot. Expected ENC values for all GC3 compositions (0-1.0) were calculated using the equation ENCexp = 2 + s + 29[s2 + (1 − s)2] − 1 [68]. When the corresponding points fall near the expected curve, mutation is the main force shaping codon usage. When the corresponding points fall considerably below the expected curve, selection is the main force shaping codon usage.
Neutrality plot mapping analysis
A neutrality plot is an analytical method used to examine the influence of mutation pressure and natural selection on the codon usage pattern [62]. In a neutrality plot, the average GC content at the first and second synonymous codon positions (GC12) are plotted against GC content at the third synonymous codon (GC3) position. The regression coefficient against GC3s is regarded as the mutation-selection equilibrium coefficient, and the evolutionary speed of the mutation pressure and natural selection pressure is expressed as the slope of a regression line. When correlation between GC12 and GC3 is statistically significant and the slope of the regression line is close to one, mutation bias is assumed to be the main force shaping codon usage. Alternatively, if the slope of the regression line is close to zero, then selection is the dominant factor.
General average hydropathicity (GRAVY), aromaticity (Aromo), and length of amino acids (L_aa)
General average hydropathicity (GRAVY) values represent the sum of the hydropathy values of all amino acids in the gene product divided by the number of residues in the sequence [37]. Aromo values denote the frequency of aromatic amino acids (Phe, Tyr, Trp) in the translated gene product [8]. The length of amino acids (L_aa) represents the number of translatable codons [21].
Correspondence analysis of RSCU
Correspondence analysis (CoA) is widely used to identify major sources of variation in synonymous codon usage among genes. COA was performed on the RSCU value using the program CodonW (http://sourceforge.net/projects/codonw) and was plotted in a 59-dimensional hyperspace according to the usage of the 59 synonymous codons. Each dimension corresponds to the RSCU value of one sense codon (excluding ATG, TGG, and stop codons). Major trends within this dataset can be determined using measures of relative inertia and genes ordered according to their positions along the axis of major inertia.
Statistical analysis
CodonW1.4.4 software was used to analyze the indices of codon usage. Correlation analyses based on Spearman’s rank correlation (with a level of significance of p < 0.05 or p < 0.01) were performed with Microsoft Excel and XLSTAT version 2014.5.03.
Results
Codon usage bias in HBoV genome
The effective codon usage statistics were used to measure the codon usage bias in the HBoV genome. The ENC values of different HBoV isolates ranged from 45.98 to 47.01, with a mean value of 46.33 ± 0.14 (Table 1). High ENC values (> 40) and little variation in ENC among different isolates suggest a moderate but highly conserved codon usage bias in the HBoV genome.
Composition analysis of HBoV genome
Codon usage bias is affected by the overall nucleotide composition of the genome. Base composition analysis of HBoV coding sequences showed an overall G+C content value of 41.89 ± 0.10% and A+T content of 58.11 ± 0.10%, indicating a strong compositional bias in favor of A+T. The G+C content in the HBoV coding sequence is not uniformly distributed, and is highest at the GC1 position (49.58 ± 0.07%), intermediate at the GC2 position (41.81 ± 0.05), and lowest at the GC3 position (34.25 ± 0.13). Further analysis of third-position wobble nucleotides revealed a significantly higher abundance of A3s (46.80%) and T3s (41.30%) than C3s (24.80%) and G3s (15.60%) (Table 1). The overall and third-position nucleotide composition values suggest that compositional constraints affect the codon usage pattern of the HBoV genome and that A/T occurs most frequently at the third codon position in HBoV genomes.
Relative synonymous codon usage (RSCU) in the HBoV genome
To investigate the patterns of synonymous codon usage and to what extent A/T-ending codons might be preferred, the relative synonymous codon usage (RSCU) was computed for each codon in the HBoV complete coding sequence and individual genes (Table 2). Among the 59 synonymous codons, 25 were identified as preferred codons (RSCU > 1.0), 21 of which were A/T-ending. On the other hand, GC-ending codons were infrequently found in the HBoV genome, with 17 out of 26 rarely used codons (RSCU value, < 0.6) being GC-ending. Further analysis revealed that the bulk of amino acids in HBoV proteins were encoded by A/T-ending codons. For example, three amino acids, arginine, serine and leucine, are encoded by six different codons, but in HBoV proteins, approximately 69% of arginine, 68% of serine, and 71% of leucine residues were encoded by A/T-ended codons. Other notable preferentially used synonymous codons for the corresponding amino acids were AAA, encoding Lys (79.34%); TTT, encoding Phe (73.18%); TAC, encoding Tyr (67.93%); GAA, encoding Glu (67.8%); GGA, encoding Gly (67.60%); CAA, encoding Gln (62.91%); and TGT, encoding Cys (60.40%).
The codon usage pattern of viruses is affected by the codon usage pattern of its host [73]. Studies have shown a great deal of variation among different viruses, ranging from complete co-incidence to complete antagonism, and in between [10, 16, 29, 45, 53, 67, 73]. Comparative analysis of the RSCU values indicated that the codon usage pattern of HBoV is mostly antagonistic to its host, with only 4-6 preferred codons coinciding with those of the host in different HBoV proteins (Supplementary Table S2). A similar mix of codon usage has been reported for several other viruses [29, 40] and this may serve to balance the translation efficiency and proper folding of viral proteins in infected host cells.
The effects of mutational biases and natural selection on synonymous codon usage
Base composition analysis of individual genes (NS1, NP1 and VP1 and VP2) revealed a similar overall G+C composition (41.10-43.80%) in all four genes (Table 1). However, the GC content at the first, second and third codon position was variable. In the NP1 gene, the GC content at all codon positions was the same, whereas in the NS1 and VP1/2 genes, the GC content was highest at the first codon position, intermediate at the second codon position, and lowest at the third codon position. Contrary to intergenomic homogeneity in the codon usage bias, analysis of individual genes revealed marked variation in ENC values. Since the VP1 and VP2 proteins differ only in the N-terminal region of VP1, no notable difference in nucleotide composition or codon usage indices were found between these two proteins. The average ENC values in different HBoV genes ranged from 41.02 to 47.92, with the highest value for NP1 (47.92 ± 0.25), an intermediate value for NS1 (46.56 ± 0.18) and the lowest values for VP1 (41.95 ± 0.22) and VP2 (41.02 ± 0.298).
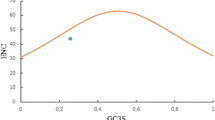
Compositional analysis revealed some notable differences in the GC3 content and codon usage bias among different HBoV genes. To elucidate the relationship between nucleotide composition and codon bias in HBoV genes, the ENC values of all four genes were plotted against the corresponding GC3s. The solid line in Figure 1A represents the curve that would be expected if codon usage were only determined by the GC content at the third codon position. In Figure 1A, data points representing individual HBoV genes formed four distinct clusters below the expected curve, and the proximity of each cluster to the standard curve differed. Data points corresponding to NS1 genes were aggregated closer to the expected curve than those of the NP1, VP1 and V2 genes, indicating a stronger influence of mutational pressure in the NS1 gene than in other genes.

NC plot and neutrality plot for 624 genes of HBoV. (A) Relationship between GC3 and the effective number of codons (ENC). The ENC values of each gene were plotted against the corresponding GC3s. The standard curve indicates the expected codon usage if GC compositional constraints alone account for codon usage bias. (B) Neutrality plot analysis of the average GC content in the first and second positions of the codons (GC12) and the GC content in the third position (GC3). The dotted line is the linear regression of GC12 against the corresponding GC3
Although the ENC–GC3 plot reflected the main factors that influenced codon usage bias, it did not indicate whether mutation pressure or natural selection was more important. To determine the extent of the role of these two evolutionary forces on the codon usage pattern of HBoV genes, we performed correlation analysis between GC content at the first and second codon positions (GC12) and that at synonymous third codon positions (GC3s).
When GC3 was plotted against GC12 for all genes (overall), a highly significant negative correlation between GC3 and GC12 was observed (r = -.899, p < 0.001), suggesting that codon usage bias may be the result of mutational pressure. However, when GC3 values of individual genes were plotted against the corresponding GC12 values, the only significant correlation between GC3 and GC12 was observed in the NS1 gene (r = 0.575, p < 0.0001) (Fig. 1B). The slope of the neutrality plot indicated that ~ 20% of the codon usage bias in the NS1 gene is due to directional mutational pressure, while ~ 80% is due to natural selection. No correlation between GC3 and GC12 was observed in NP1 (r = 0.026, p = 0.750), VP1 (r = 0.042, p = 0.602) and VP2 (r = -0.037, p = 0.6426) genes. The slopes of the regression lines in NP1, VP1/2 were calculated to be 0.004 and 0.008, respectively, demonstrating the dominant influence of natural selection. Taken together; these data suggest that different evolutionary pressures are acting disproportionally on HBoV genes. Mutational pressure emerged as a minor factor acting on the NS1 gene only, while natural selection emerged as the dominant factor influencing the codon usage bias in all HBoV genes.
Effect of gene expression level on synonymous codon usage bias
To compare the codon usage preference with respect to its host, CAI values for all genes were calculated using the human codon usage as a reference (Table 1). The CAI for HBoV genes varied from 0.701 to 0.767, with a mean of 0.731 ± 0.001, which indicates that most of the HBoV genes are highly expressive in nature. Among the four HBoV genes, NP1 has the highest CAI value (0.768 ± 0.002), followed by capsid proteins VP1 (0.753 ± 0.002) and VP2 (0.742 ± 0.0002). The NS1 (0.701 ± 0.001) has the lowest CAI value among all HBoV proteins. To assess the effect of gene expression on codon usage bias, correlation between the ENC and CAI of each gene was determined. A significant positive correlation between ENC and CAI in the VP1 and VP2 genes (r = 0.260, p < 0.001 and r = 0.261, p < 0.001, respectively) indicates that codon usage in these genes is affected by gene expression level. No direct correlation between ENC and CAI was found for the NS1 and NP1 genes.
Relationship between codon bias and protein length, hydropathicity index, and aromaticity score
Physical and chemical properties of proteins, such as hydropathicity, aromaticity and length of the encoded protein play important roles in shaping codon usage of some species [44, 52]. In order to investigate the potential role of hydropathicity, aromaticity, and protein length in HBoV codon usage bias, correlation coefficients between ENC, hydropathicity index (Gravy), aromaticity score (Aromo) and protein length (L_aa) were determined (Table S3). Protein length was significantly correlated with codon usage bias (-0.591, p < 0.0001). A significant negative correlation between GRAVY and ENC (NS1: r = -0.210, p < 0.01), GRAVY and axis 1 (NP1: r = - 0.181, p < 0.05, VP1 r = -0.176, p < 0.05) and a positive correlation between Aromo and ENC (NS1: r = 0.326, p < 0.001) suggest that both hydropathicity and aromaticity significantly influence the codon usage bias in HBoV genes.
Correspondence analysis
To investigate synonymous codon usage variation among HBoV genes, COA was implemented for all 624 genes from 156 HBoV isolates. Figure 2A depicts the position of each ORF on the plane defined by the first and second principal axes generated by COA on RSCU values of genes. The first and second principal axes account for 61.24% and 35.46% of the total variability, indicating that first two axes are major explanatory axes for interpreting codon usage variation among genes.

Correspondence analysis of synonymous codon usage in HBoV genome. (A) The relative and cumulative inertia of the first 40 factors from a COA of the relative synonymous codon usage values. (R.Iner., relative inertia; R.Sum, relative sum or cumulative relative inertia). (B) distribution of HBoV genes on the primary and secondary axes
The plot of the first and second axes of each gene is shown in Figure 2B. The distribution of HBoV genes based on RSCU values on first and second axes resulted in four distinct clusters, each representing one HBoV gene. Genes falling in the same cluster indicate that these genes have more or less similar codon usage biases. Further analysis revealed the separation of genes along the first and second axes based on codon usage bias (ENC) and gene expression level (CAI). It was noticeable from Figure 2B that genes with low codon usage bias (ENC > 46; NS1 = 46.58 and NP1 = 47.94) were located to the positive side of axis 1, while genes with high codon usage bias (ENC < 42; VP1 = 41.95 and VP2 = 41.02) were located to the negative side of axis 1. On other hand, genes with low expression levels (CAI < 0.71; NS1 = 0.70) were located to the negative side of axis 2, while genes with high expression levels (CAI > 0.74; NP1 = 0.768, VP1 = 0.753 and VP2 = 0.742) were located to the positive side of the axis 2. A highly significant positive correlation between CAI (overall), principle axis 1 and principle axis 2, (r = 0.249, p < 0.0001; and r = 0.949, p < 0.0001, respectively) suggest that codon usage in HBoV is influenced by gene expression levels.
Dinucleotide frequency
Codon usage can also be influenced by underlying biases in dinucleotide frequency, which differs greatly among organisms. To investigate the potential effect of dinucleotide bias on codon usage, relative the abundance of the 16 dinucleotides in coding and non-coding HBoV sequences was calculated. The ratio of the observed to the expected frequency for each dinucleotide in the entire coding sequence (CDS), non-coding sequence, and all four genes is shown in Figure 3.

Relative dinucleotide abundance in the HBoV genome. The scatter plot represents the observed/expected (O/E) frequency ratio of 16 dinucleotides in each gene. The mean ± standard deviation of dinucleotide O/E ratios for HBoV CDS is 1.0 ± 0.246. The mean ± standard deviation of dinucleotide O/E ratios for HBoV NS1, NP1, VP1 and VP2 are 1.0 ± 0.23, 1.0 ± 0.273, 1.0 ± 0.284 and 1.0 ± 0.310, respectively. The solid line represents the O/E of 16 dinucleotides in the coding region, (CDS) and a dotted line represent the O/E ratio of 16 dinucleotides in the non-coding region of the HBoV genome. A dotted box indicates the normal range. Insets: under- and over-represented dinucleotides in each gene
The mean ± standard deviation of dinucleotide O/E ratios for the HBoV genome is 1.0 ± 0.246. Dinucleotides located outside the confidence interval (mean ± 1SD, 0.75–1.25) were classified as low or high relative abundance, respectively. In the coding regions of the HBoV genome, seven dinucleotides exhibit substantial deviation from the expected frequency (Fig. 3A, sold line). Out of seven dinucleotides, four nucleotide pairs (CpA, CpT, GpG and TpG) are over-represented (O/E > 1.25), whereas three nucleotide pairs (CpG, GpT and TpA) are under-represented (O/E < 0.75). In the non-coding sequence, all dinucleotides are within the threshold value (Fig. 3A, dotted line). Similar dinucleotide biases were observed in individual HBoV genes (Fig. 3B-E). In the NS1 gene, three dinucleotides (CpT, GpG and TpG) are over-represented, while four nucleotide pairs (CpC, CpG, GpT and TpA) are suppressed. In the NP1 gene, two nucleotide pairs (CpT and TpC) are over-represented, while three nucleotide pairs (CpG, GpT and TpA) are under-represented. In VP1 and VP2, three nucleotide pairs (CpA, GpG and TpG,) are over-represented and two nucleotide pairs (CpG and TpA) are under-represented.
Among the differentially used dinucleotides, only CpG and TpA depletion was common across all four HBoV genes. We also observed a slight decrease in CpGO/E and TpAO/E ratio (0.776 and 0.86, respectively) in the non-coding sequence, which indicates that compositional constraint is a contributing factor in CpG and TpA depletion, but the extent of CpG and TpA depletion in the coding region cannot be explained by compositional constraint alone. To determine the effect of evolutionary pressures other than compositional constraint, O/E ratios for CpG/GpC and TpA/ApT were calculated. A high O/E ratio (~ 1.0) would indicate that depletion was due to pressure acting upon constituent nucleotides, whereas a low O/E ratio would suggest evolutionary pressures acting specifically upon dinucleotides. The CpG/GpCO/E and TpA/ApTO/E ratios of all four genes (NS1 = 0.45 & 0.94, NP1 = 0.46 & 0.70, VP1 = 0.31 & 0.67 and VP2 = 0.26 & 0.63, respectively) suggest that CpG and TpA depletion in HBoV genes is a combined effect of pressures acting on the constituent mononucleotides and evolutionary pressures acting specifically on CpG and TpA dinucleotides.
Depletion or over-representation of dinucleotides was expected to affect not only synonymous codon preferences but also amino acid frequencies and the choice of the synonymous codons. To investigate the possible effects of CpG and TpA depletion on codon usage bias, the RSCU value of the eight CpG-containing codons (ACG, CCG, GCG, TCG, CGA, CGC, CGG and CGT) and six TpA-containing codons (ATA, CTA, GTA, TTA, TAC and TAT) were analyzed in all four genes. In NS1, six out of 14 codons (CGG [0.00], ACG [0.00], TCG [0.18], CGA [0.23], GCG [0.30] and CCG [0.34]) were markedly suppressed, four codons (GTA [0.57], ATA [0.82], TAT [0.91] and CGT [0.92]) were slightly suppressed, and four codons (TAC [1.09], TTA [1.17], CTA [1.31] and CGC [1.30]) were overused. In the NP1 gene, six codons (CGG [0.00], GCG [0.00], ACG [0.29], TAT [0.29], CGT [0.29], and CGA [0.29]) were markedly suppressed, five codons (CCG [0.68], TTA [0.86], CGC [0.86], ATA [0.86], and TCG [0.89]) were slightly underused, and three codons (CTA [3.43], GTA [1.08] and TAC [1.71]) were overused. In the VP1 and VP2 genes, the effect of CpG and TpA depletion was more pronounced, and 10 (TCG, CGG, CGA, GCG, CCG, ACG, TTA, CGT, TAT and ATA) out of 14 codons were underused. Taken together, these data suggest that depletion of CpG and TpA dinucleotides in HBoV affects the codon usage.
Discussion
Our analysis revealed that HBoV lacks strong codon usage bias, which is consistent with previously reported data on human bocavirus and other parvoviruses [58, 72]. Weak codon bias seems to be an adaptive strategy employed by a large number of RNA and DNA viruses [31, 54], which may enable these viruses to maintain efficient replication without competing for limited tRNA resources. In accordance with previously published data, the pattern of codon usage in different HBoV proteins was mostly antagonistic to its host, sharing only 3-6 preferred codons, which may help the virus to maintain steady replication using non-optimal codons. Although codon usage bias appears to be conserved between HBoV strains, significant intragenomic variation in codon usage has been observed. The structural genes exhibited a higher degree of codon usage bias (ENC < 42) than the non-structural genes (ENC > 46). A marked difference in codon usage between the structural and non-structural genes suggests that HBoV genes have taken different codon evolution pathways depending on the function of each protein. Non-structural proteins are required for early stages of the viral life cycle, and low codon usage bias in these proteins would allow a less restricted tRNA preference and more-efficient viral RNA replication. On the other hand, structural proteins are the main target of the humoral immune response and are often required at the late stage of the viral life cycle; therefore, more biased codon usage may allow a more gradual accumulation of these proteins without being detected by the host immune response.
Natural selection and mutation pressure are thought to be two main factors that account for codon usage variation in different organisms. A general association between codon usage bias and base composition in the HBoV genome suggests that mutational pressure contributes significantly to the codon usage bias. However, a significant distance between the expected and actual ENC values in all HBoV genes and a lack of significant correlation between GC12 and GC3 in the HBoV NP1 and VP1 and VP2 genes suggest that natural selection is the main factor that determines the codon usage bias in HBoV. These findings are in agreement with results published for 58 parvoviruses [58] but in disagreement with results published for 11 human bocavirus strains [72]. Additionally, significant correlations between ENC and gene length, CAI (VP1/2), Gravy (NS1) and Aromo (NS1) suggest that HBoV codon bias is multifactorial and that different evolutionary pressures are acting disproportionally on HBoV genes.
Codon usage bias can also be influenced by dinucleotide frequency. It has been reported that the proportion of codon usage bias due to mutation pressure in RNA virus often increases after accounting for dinucleotide bias [31]. The O/E frequency ratio of the 16 possible dinucleotides in the HBoV coding region revealed that ~ 40% of dinucleotides were outside of the normal range (< 0.75, > 1.25) and can be classified as under- or over-represented. The frequency of occurrence of 16 dinucleotides in individual HBoV genes was slightly variable, with CpG and TpA dinucleotides suppressed in almost all gene. Comparative analysis of CpG and TpA dinucleotides frequencies in coding and non-coding regions revealed a significant depletion of CpG and TpA in coding regions compared to non-coding regions, suggesting a prevailing influence of translation selection shaping the relative abundance of specific dinucleotides. A low prevalence of CpG dinucleotides has been observed in the genome of many vertebrates and DNA and RNA viruses [4, 34]. Several possible reasons, such as lower transcription rate for CpG-containing codons, stimulation of Toll-like-receptor-mediated innate immune response by unmethylated CpGs, and spontaneous deamination of methylated cytosine in CpG dinucleotides have been suggested to play a role in the depletion of CpG dinucleotides [12, 30, 43]. The CpG motif in A/U-rich RNA has immunostimulatory properties, and its frequency in influenza virus has decreased over the course of several decades of human adaptation [23]. An experimental increase in CpG and UpA dinucleotides in echovirus 7 has been shown to have a negative impact on replication kinetics [22]. UpA deficiency has also been reported in some other RNA viruses [65]. UpA dinucleotides in cytoplasmic mRNA are under direct selection, as the dinucleotide is recognized by RNA-degrading enzymes in the cytoplasm. The UpA composition therefore modulates protein expression from mRNA through its influence on transcriptome turnover [6, 20]. Marked suppression of CpG and UpA dinucleotides in HBoV mRNA sequences may therefore reflect selection for increased stability of viral mRNA in the cytoplasm and reduced immunostimulatory effects.
In summary, our analyses revealed a moderate but highly conserved gene-specific codon usage bias in the HBoV genome. The codon usage bias in the HBoV genome is mainly affected by natural selection, whereas mutational pressure emerged as a minor contributing factor. Dinucleotide bias also emerged as a key determinant of codon usage bias. The CpG/TpA dinucleotides are depleted across all HBoV genes, and depletion of dinucleotides was found to be a combined effect of compositional constraints imposed by nucleotide composition of the genome and evolutionary pressures specifically acting on CpG and TpA dinucleotides. Gene length, gene expression, and protein properties emerged as minor factors and contributed to codon usage bias in some HBoV genes. The results of this study not only elucidate the mechanisms underlying the molecular evolution of HBoV but also provide a theoretical basis for improving HBoV gene expression by codon optimization and for studying the functional and pathological relevance of various HBoV proteins. Furthermore, this study may also provide a foundation for the development of a live attenuated vaccine strain containing thousands of silent mutations, which would present a significant barrier against reversion.
References
Akashi H (1997) Codon bias evolution in Drosophila. Population genetics of mutation-selection drift. Gene 205:269–278
Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B (2005) Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci USA 102:12891–12896
Arthur JL, Higgins GD, Davidson GP, Givney RC, Ratcliff RM (2009) A novel bocavirus associated with acute gastroenteritis in Australian children. PLoS Pathog 5:e1000391
Atkinson NJ, Witteveldt J, Evans DJ, Simmonds P (2014) The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res 42:4527–4545
Bernardi G, Bernardi G (1986) Compositional constraints and genome evolution. J Mol Evol 24:1–11
Beutler E, Gelbart T, Han JH, Koziol JA, Beutler B (1989) Evolution of the genome and the genetic code: selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc Natl Acad Sci USA 86:192–196
Bradel-Tretheway BG, Zhen Z, Dewhurst S (2003) Effects of codon-optimization on protein expression by the human herpesvirus 6 and 7 U51 open reading frame. J Virol Methods 111:145–156
Brocchieri L, Karlin S (1994) Geometry of interplanar residue contacts in protein structures. Proc Natl Acad Sci USA 91:9297–9301
Bulmer M (1991) The selection-mutation-drift theory of synonymous codon usage. Genetics 129:897–907
Butt AM, Nasrullah I, Tong Y (2014) Genome-wide analysis of codon usage and influencing factors in chikungunya viruses. PLoS One 9:e90905
Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH (2004) Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci USA 101:3480–3485
Chinnery HR, McLenachan S, Binz N, Sun Y, Forrester JV, Degli-Esposti MA, Pearlman E, McMenamin PG (2012) TLR9 ligand CpG-ODN applied to the injured mouse cornea elicits retinal inflammation. Am J Pathol 180:209–220
Chow BD, Ou Z, Esper FP (2010) Newly recognized bocaviruses (HBoV, HBoV2) in children and adults with gastrointestinal illness in the United States. J Clin Virol 47:143–147
Costafreda MI, Perez-Rodriguez FJ, D’Andrea L, Guix S, Ribes E, Bosch A, Pinto RM (2014) Hepatitis A virus adaptation to cellular shutoff is driven by dynamic adjustments of codon usage and results in the selection of populations with altered capsids. J Virol 88:5029–5041
Cotmore SF, Agbandje-McKenna M, Chiorini JA, Mukha DV, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D, Davison AJ (2014) The family Parvoviridae. Arch Virol 159:1239–1247
Cristina J, Moreno P, Moratorio G, Musto H (2015) Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res 196:87–93
D’Onofrio G, Ghosh TC, Bernardi G (2002) The base composition of the genes is correlated with the secondary structures of the encoded proteins. Gene 300:179–187
Deng ZH, Hao YX, Yao LH, Xie ZP, Gao HC, Xie LY, Zhong LL, Zhang B, Cao YD, Duan ZJ (2014) Immunogenicity of recombinant human bocavirus-1,2 VP2 gene virus-like particles in mice. Immunology 142:58–66
Dittmar KA, Goodenbour JM, Pan T (2006) Tissue-specific differences in human transfer RNA expression. PLoS Genet 2:e221
Duan J, Antezana MA (2003) Mammalian mutation pressure, synonymous codon choice, and mRNA degradation. J Mol Evol 57:694–701
Duret L, Mouchiroud D (1999) Expression pattern and surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci USA 96:4482–4487
Fros JJ, Dietrich I, Alshaikhahmed K, Passchier TC, Evans DJ, Simmonds P (2017) CpG and UpA dinucleotides in both coding and non-coding regions of echovirus 7 inhibit replication initiation post-entry. eLife 6:e29112
Greenbaum BD, Ghedin E (2015) Viral evolution: beyond drift and shift. Curr Opin Microbiol 26:109–115
Gurda BL, Parent KN, Bladek H, Sinkovits RS, DiMattia MA, Rence C, Castro A, McKenna R, Olson N, Brown K, Baker TS, Agbandje-McKenna M (2010) Human bocavirus capsid structure: insights into the structural repertoire of the parvoviridae. J Virol 84:5880–5889
Haas J, Park EC, Seed B (1996) Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr Biol 6:315–324
Han TH, Chung JY, Hwang ES (2009) Human bocavirus 2 in children, South Korea. Emerg Infect Dis 15:1698–1700
Hershberg R, Petrov DA (2008) Selection on codon bias. Annu Rev Genet 42:287–299
Hooper SD, Berg OG (2000) Gradients in nucleotide and codon usage along Escherichia coli genes. Nucleic Acids Res 28:3517–3523
Hu JS, Wang QQ, Zhang J, Chen HT, Xu ZW, Zhu L, Ding YZ, Ma LN, Xu K, Gu YX, Liu YS (2011) The characteristic of codon usage pattern and its evolution of hepatitis C virus. Infect Genet Evol 11:2098–2102
Jabbari K, Bernardi G (2004) Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 333:143–149
Jenkins GM, Holmes EC (2003) The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92:1–7
Kapoor A, Simmonds P, Slikas E, Li L, Bodhidatta L, Sethabutr O, Triki H, Bahri O, Oderinde BS, Baba MM, Bukbuk DN, Besser J, Bartkus J, Delwart E (2010) Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J Infect Dis 201:1633–1643
Kapoor A, Hornig M, Asokan A, Williams B, Henriquez JA, Lipkin WI (2011) Bocavirus episome in infected human tissue contains non-identical termini. PLoS One 6:e21362
Karlin S, Doerfler W, Cardon LR (1994) Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J Virol 68:2889–2897
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kunec D, Osterrieder N (2016) Codon pair bias is a direct consequence of dinucleotide bias. Cell Rep 14:55–67
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Li J, Yang Y, Dong Y, Li Y, Huang Y, Yi Q, Liu K, Li Y (2013) Key elements of the human bocavirus type 1 (HBoV1) promoter and its trans-activation by NS1 protein. Virol J 10:315
Li Q, Zhang Z, Zheng Z, Ke X, Luo H, Hu Q, Wang H (2013) Identification and characterization of complex dual nuclear localization signals in human bocavirus NP1: identification and characterization of complex dual nuclear localization signals in human bocavirus NP1. J Gen Virol 94:1335–1342
Liu YS, Zhou JH, Chen HT, Ma LN, Pejsak Z, Ding YZ, Zhang J (2011) The characteristics of the synonymous codon usage in enterovirus 71 virus and the effects of host on the virus in codon usage pattern. Infect Genet Evol 11:1168–1173
Luo H, Zhang Z, Zheng Z, Ke X, Zhang X, Li Q, Liu Y, Bai B, Mao P, Hu Q, Wang H (2013) Human bocavirus VP2 upregulates IFN-beta pathway by inhibiting ring finger protein 125-mediated ubiquitination of retinoic acid-inducible gene-I. J Immunol 191:660–669
Lusebrink J, Schildgen V, Tillmann RL, Wittleben F, Bohmer A, Muller A, Schildgen O (2011) Detection of head-to-tail DNA sequences of human bocavirus in clinical samples. PLoS One 6:e19457
Medvedeva YA, Fridman MV, Oparina NJ, Malko DB, Ermakova EO, Kulakovskiy IV, Heinzel A, Makeev VJ (2010) Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genom 11:48
Moriyama EN, Powell JR (1998) Gene length and codon usage bias in Drosophila melanogaster, Saccharomyces cerevisiae and Escherichia coli. Nucleic acids Res 26:3188–3193
Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E (2006) Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol 80:9687–9696
Ngumbela KC, Ryan KP, Sivamurthy R, Brockman MA, Gandhi RT, Bhardwaj N, Kavanagh DG (2008) Quantitative effect of suboptimal codon usage on translational efficiency of mRNA encoding HIV-1 gag in intact T cells. PLoS One 3:e2356
Novembre JA (2002) Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol 19:1390–1394
Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12:32–42
Puigbo P, Bravo IG, Garcia-Vallve S (2008) CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3:38
Qiu J, Soderlund-Venermo M, Young NS (2017) Human parvoviruses. Clin Microbiol Rev 30:43–113
Qu XW, Liu WP, Qi ZY, Duan ZJ, Zheng LS, Kuang ZZ, Zhang WJ, Hou YD (2008) Phospholipase A2-like activity of human bocavirus VP1 unique region. Biochem Biophys Res Commun 365:158–163
Romero H, Zavala A, Musto H (2000) Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic acids Res 28:2084–2090
Sanchez G, Bosch A, Pinto RM (2003) Genome variability and capsid structural constraints of hepatitis a virus. Journal of virology 77:452–459
Shackelton LA, Parrish CR, Holmes EC (2006) Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol 62:551–563
Sharp CP, LeBreton M, Kantola K, Nana A, Diffo Jle D, Djoko CF, Tamoufe U, Kiyang JA, Babila TG, Ngole EM, Pybus OG, Delwart E, Delaporte E, Peeters M, Soderlund-Venermo M, Hedman K, Wolfe ND, Simmonds P (2010) Widespread infection with homologues of human parvoviruses B19, PARV4, and human bocavirus of chimpanzees and gorillas in the wild. J Virol 84:10289–10296
Sharp PM, Li WH (1986) An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol 24:28–38
Sharp PM, Li WH (1987) The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15:1281–1295
Shi SL, Jiang YR, Liu YQ, Xia RX, Qin L (2013) Selective pressure dominates the synonymous codon usage in parvoviridae. Virus Genes 46:10–19
Simmonds P (2012) SSE: a nucleotide and amino acid sequence analysis platform. BMC Res Notes 5:50
Smith DW (1996) Problems of translating heterologous genes in expression systems: the role of tRNA. Biotechnol Prog 12:417–422
Soderlund-Venermo M, Lahtinen A, Jartti T, Hedman L, Kemppainen K, Lehtinen P, Allander T, Ruuskanen O, Hedman K (2009) Clinical assessment and improved diagnosis of bocavirus-induced wheezing in children, Finland. Emerg Infect Dis 15:1423–1430
Sueoka N (1988) Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci USA 85:2653–2657
Sun B, Cai Y, Li Y, Li J, Liu K, Li Y, Yang Y (2013) The nonstructural protein NP1 of human bocavirus 1 induces cell cycle arrest and apoptosis in Hela cells. Virology 440:75–83
Tewary SK, Zhao H, Shen W, Qiu J, Tang L (2013) Structure of the NS1 protein N-terminal origin recognition/nickase domain from the emerging human bocavirus. J Virol 87:11487–11493
Tulloch F, Atkinson NJ, Evans DJ, Ryan MD, Simmonds P (2014) RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. eLife 3:e04531
Wang M, Liu YS, Zhou JH, Chen HT, Ma LN, Ding YZ, Liu WQ, Gu YX, Zhang J (2011) Analysis of codon usage in Newcastle disease virus. Virus Genes 42:245–253
Wang M, Zhang J, Zhou JH, Chen HT, Ma LN, Ding YZ, Liu WQ, Liu YS (2011) Analysis of codon usage in bovine viral diarrhea virus. Arch Virol 156:153–160
Wright F (1990) The ‘effective number of codons’ used in a gene. Gene 87:23–29
Zhang Z, Zheng Z, Luo H, Meng J, Li H, Li Q, Zhang X, Ke X, Bai B, Mao P, Hu Q, Wang H (2012) Human bocavirus NP1 inhibits IFN-beta production by blocking association of IFN regulatory factor 3 with IFNB promoter. J Immunol 189:1144–1153
Zhao B, Yu X, Wang C, Teng Z, Wang C, Shen J, Gao Y, Zhu Z, Wang J, Yuan Z, Wu F, Zhang X, Ghildyal R (2013) High human bocavirus viral load is associated with disease severity in children under five years of age. PLoS One 8:e62318
Zhao KN, Gu W, Fang NX, Saunders NA, Frazer IH (2005) Gene codon composition determines differentiation-dependent expression of a viral capsid gene in keratinocytes in vitro and in vivo. Mol Cell Biol 25:8643–8655
Zhao S, Zhang Q, Liu X, Wang X, Zhang H, Wu Y, Jiang F (2008) Analysis of synonymous codon usage in 11 human bocavirus isolates. Bio Syst 92:207–214
Zhou H, Wang H, Huang LF, Naylor M, Clifford P (2005) Heterogeneity in codon usages of sobemovirus genes. Arch Virol 150:1591–1605
Zhou J, Liu WJ, Peng SW, Sun XY, Frazer I (1999) Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability. J Virol 73:4972–4982
Acknowledgements
We thank the College of Clinical Pharmacy for providing necessary support to conduct this research. This research was supported by the Deanship of Scientific Research, King Faisal University, grant #160009.
Author information
Authors and Affiliations
Contributions
SH, STR and AHA identified the research topic and designed the study. SH collected the data and conducted the analysis. SH, STR and AHA prepared the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This research work does not involve human participants or animals.
Informed consent
Not applicable, as no human participants are involved.
Additional information
Handling Editor: Sheela Ramamoorthy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hussain, S., Rasool, S.T. & Asif, A.H. A detailed analysis of synonymous codon usage in human bocavirus. Arch Virol 164, 335–347 (2019). https://doi.org/10.1007/s00705-018-4063-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-4063-8