
Abstract
Autophagy is a homeostatic process that has been shown to be vital in the innate immune defense against pathogens. However, little is known about the regulatory role of autophagy in porcine teschovirus 2 (PTV-2) replication. In this study, we found that PTV-2 infection induces a strong increase in GFP-LC3 punctae and endogenous LC3 lipidation. However, PTV-2 infection did not enhance autophagic protein degradation. When cellular autophagy was pharmacologically inhibited by wortmannin or 3-methyladenine, PTV-2 replication increased. The increase in virus yield via autophagy inhibition was further confirmed by silencing atg5, which is required for autophagy. Furthermore, PTV-2 replication was suppressed when autophagy was activated by rapamycin. Together, the results suggest that PTV-2 infection activates incomplete autophagy and that autophagy then inhibits further PTV-2 replication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine teschovirus (PTV) is a non-enveloped, positive-sense, single-stranded RNA virus of 25-30 nm in diameter, classified in the genus Teschovirus within the family Picornaviridae [1]. It comprises 13 known serotypes to date [2, 3]. PTVs have contaminated pig herds worldwide (endemic or enzootic) together with a variety of common swine pathogens [4], and could be used as potential indicator viruses for fecal contamination during pork carcass processing [5]. Highly virulent strains of PTV-1 are known to cause teschovirus encephalomyelitis. PTV-2, -3, and -5 are associated with Talfan disease (a milder presentation of polioencephalomyelitis) [6].
Autophagy is an important cellular homeostatic process that can contribute to innate and adaptive immunity through the clearing of intracellular pathogens. While positive-stranded RNA viruses have been shown to hijack this process for their own benefit [7], autophagy can act as a host defense mechanism against some viruses, such as Sindbis virus [8] and herpes simplex virus [9], by removing them from cells.
Autophagy is already known to have a key role in picornaviral infections [10]. For example, poliovirus and coxsackie virus B3 utilize the autophagic machinery to facilitate virus production [11]. However, it remains unclear whether autophagy is involved in PTV infection. In this study, we sought to clarify whether PTV-2 infection induces autophagy, and if it does, how does cellular autophagy affect virus replication. We report that PTV-2 infection triggers incomplete autophagy and that autophagy could limit PTV-2 replication in PK-15 cells.
Materials and methods
Cells and virus
The PK-15 cell line, free of porcine circovirus contamination, was used. The PK-15 and PK-15/EGFP-LC3 cells (the PK-15 cell line stably expressing EGFP-LC3) [12] were cultured in Dulbecco’s minimal essential medium (HyClone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/ml penicillin G, and 100 μg/ml streptomycin (Gibco). The cells were cultured at 37 °C in a 5% CO2 incubator.
The PTV-2 isolate ZJ16LX0401 strain (GenBank ID: KX527849) was isolated in Zhejiang, China, and was purified by endpoint dilution assay and propagated on the PK-15 cell monolayer. To obtain the replication-incompetent PTV-2, 5 ml of virus suspension were dispensed to form a layer of fluid in an open, cell culture dish, on ice, and were irradiated with a UV lamp (UVP LLC, Upland, CA, USA) at a wavelength of 254 nm for 3 h at approximately 10 cm from the light source. The absence of virus infectivity after UV treatment was confirmed by TCID50 assay, as described below.
Virus infection and treatments with chemicals
The PK-15 or PK-15/EGFP-LC3 cells were infected with PTV-2 at a multiplicity of infection (MOI) of 0.1. Infection was allowed to proceed for 1 h and the supernatant was then removed. For analysis of autophagic flux, PK-15 cells were mock-infected or infected with PTV-2 for 9 h. Subsequently, the cells were treated for 6 h in the absence or presence of 5 μM chloroquine (CQ, Selleck, Houston, TX, USA) before the cells were then lysed for Western blotting of LC3-II and p62/SQSTM1 (p62). For autophagy alteration, PK-15 or PK-15/EGFP-LC3 cells were pretreated with 2 μM mTOR inhibitor rapamycin (Rapa, Selleck), 5 mM 3-methyladenine (3-MA, Sigma-Aldrich, St. Louis, MO, USA) or 1 μM Wortmannin (WM, Selleck) for 10 h (both as phosphoinositide 3-kinase (PI3 K) inhibitor) at 37 °C. The cells were then infected, and further incubated in fresh media in the presence or absence of Rapa, 3-MA or WM at the same concentrations as for pre-treatments, or the corresponding concentration of the solvent dimethyl sulfoxide (DMSO, control).
siRNA and transfection
For Atg5 silencing, siAtg5 was transfected into PK-15 cells with lipofectamine 2000 (Invitrogen, Eugene, OR, USA) as per the manufacturer’s instructions. Cells were infected with PTV-2 24 h post-transfection, and then incubated for 24 h. The infected cells were then collected for further analyses. The Atg5 knockdown efficiency was evaluated by Western blotting. The siRNA against Atg5 and control scrambled siRNA (siCtrl) were purchased from Genepharma (Shanghai, China). The sense strand of siAtg5 is 5′-GGAUGUAAUUGAAGCUCAU-3′.
Western blotting
After treatment or transfection for the indicated times, all samples were washed with ice-cold PBS and lysed for 10 min in ice-cold lysis buffer (50 mM Tris-HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40 with complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany)). Cell debris were pelleted by centrifugation at 12,000 rpm, 4 °C for 20 min, and the supernatants were transferred to fresh tubes kept on ice. Protein concentration was measured using the BCA protein assay kit (MultiSciences, Hangzhou, China). Protein samples were boiled at 100 °C for 5 min in the presence of SDS-PAGE loading buffer. Approximately 30 μg of total protein from cell lysates was loaded into the wells of a 12% SDS-PAGE gel. The gel was run for 1-2 h at 100 V and then transferred onto PVDF membrane. The blots were blocked for 1 h at room temperature in Tris-buffered saline (25 mM Tris pH 7.5, 150 mM NaCl) containing 0.05% Tween 20 and 5% nonfat milk, and then incubated overnight at 4 °C with the following primary antibodies: mouse anti-Atg5 monoclonal IgG (Sigma-Aldrich, A2859, 1:1000), rabbit anti-p62 (Sigma-Aldrich, P0067, 1:1000) or LC3 polyclonal IgG (Sigma-Aldrich, L7543, 1:2000), and mouse anti-β-actin monoclonal IgG (CST, Boston, MA, USA, 3700, 1:1000). The blots were washed five times with TBST and incubated for 1 h at 37 °C with goat anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase-labeled (HRP) (KPL, Gaithersburg, MD, USA). The bands were visualized using SuperSignal West Pico chemiluminescent substrate (Thermo, Marina, CA, USA) with a Gel 3100 Chemiluminescent and Fluorescent Imaging System (Sagecreation, Beijing, China). All antibodies were found to react with target molecules from porcine cell lines used in this study.
Confocal microscopy
For detection of autophagosomes, PK-15/EGFP-LC3 cells, cultured in a petri dish with a glass-bottom, were infected with PTV-2 or treated with 2 μM rapamycin for the indicated periods of time. Fixed and permeabilized cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Fluorescence of EGFP-LC3 was observed by laser scanning confocal microscope (FV1000, Olympus). The average number of EGFP-LC3 punctae per cell from at least 50 cells per sample was counted (Gu et al., 2016).
For detection of degradation of autophagosomes, infected and uninfected cells were washed with PBS, fixed and permeabilized 9 h post infection. The cells were stained with anti-LAMP1 polyclonal IgG (Sigma-Aldrich, SAB3500285, 1:200), followed by a DyLightTM 549 conjugated goat anti-rabbit IgG (MultiSciences, Hangzhou). Nuclear DNA were stained with DAPI and cells were captured by confocal microscope. For staining of acidic compartments, cells were treated with 50 nM LysoTracker Red DND-99 (Invitrogen) to visualize lysosomes.
Electron microscopy
PTV-2- or mock-infected PK-15 cells were processed as previously described [13]. These samples were then analysed using a Hitachi H-9500 transmission electron microscope (Hitachi High-Technologies Corporation, Japan).
Indirect immunofluorescence and virus titration
Indirect immunofluorescence assay was performed as described previously [12]. The mouse antiserum to PTV-2 was generated by intraperitoneal injection of 105 TCID50 of virus into BALB/c mice with boosting after 21 days. Anti-PTV-2 BALB/c mice positive serum was used at a dilution of 1:500 followed by fluorescein-labeled goat anti-mouse IgG (KPL) at 37 °C for 45 min. Virus titers were calculated using the Reed-Muench method and expressed as TCID50/ml.
Quantification of viral RNA by real-time RT-PCR
The viral RNA was extracted from the cell culture supernatants of infected cells using the QIAamp Viral RNA Mini Kit (Qiagen, Beijing, China). Viral RNA level was quantified with the primers PTV-F (5′-GACAGAGTACAGAAGAGCAAGT-3′) and PTV-R (5′- TGACTATACAAAGTACAGACGG-3′) via real-time RT-PCR on a BIO-RAD iQ™5 machine (Berkeley, CA, USA). Viral RNA copy numbers were interpolated from a standard curve established by detecting the CT values of the serially diluted plasmid DNA containing the 5′-UTR sequence of PTV.
Statistical analysis
The results were shown as mean ± SD from at least three independent experiments and analyzed by using the Student t-tests.
Results
PTV-2 infection induces autophagosome formation
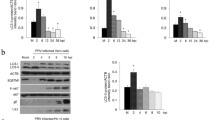
To investigate whether PTV-2 infection could trigger autophagy machinery and induce autophagosome formation, PK-15 cells were infected with PTV-2. Electron microscopy, the presence of fluorescent GFP-LC3 punctae, or LC3 lipidation were all used to monitor autophagy. Transmission electron microscopy is a valid and gold standard technique for observing autophagosomes in cells. Figure 1A shows that autophagic vesicles were observed in PTV-2-infected PK-15 cells. Correspondingly, PTV-2 or the autophagy inducer rapamycin (an mTOR pathway inhibitor) induced a significant increase in the number of autophagosome-like vesicles (EGFP-LC3 punctae) at 9 h post-infection (hpi), as compared to the uninfected control cells (Fig. 1B and C, P < 0.01). To verify whether PTV virions were the target of autophagy, PTV-2-infected PK-15/EGFP-LC3 cells were stained with anti-PTV-2 BALB/c mice positive serum. Confocal microscopy revealed that viral proteins colocalize with EGFP-LC3 punctae (Fig. 1D). To further determine whether the observed autophagosome-like vesicles were truly related to autophagy, we detected lipidated LC3 (LC3-II), another hallmark of autophagy. Western blotting showed that PTV-2-infected and rapamycin-treated cells had an increased ratio of LC3-II to β-actin, relative to uninfected cells (Fig. 1E) with statistical significance at 6 h (P < 0.05) and 9 h (P < 0.01) post-infection (Fig. 1F). Similar results were observed in PTV-2-infected swine testicle (ST) cells (Fig. S1). Therefore, these data strongly indicate that PTV-2 infection triggers autophagy in PK-15 cells, as shown by induction of LC3-II and increased formation of autophagosome-like vesicles. To assess whether cells have to be actively infected or whether inactivated viral particles are sufficient to induce autophagosome accumulation, we inoculated cells with live or UV-inactivated PTV-2 and analyzed LC3-II levels by Western blotting. We found only live PTV-2 could elicit increased level of LC3-II, whereas UV-inactivated virus did not induce observable change in LC3-II levels, indicating that live PTV-2 infection is needed for autophagy induction (Fig. S2).

PTV-2 infection induces autophagosome accumulation in PK-15 cells. (A) Autophagic vacuoles in PTV-2-infected cells observed by transmission electron microscopy. The vesicles with characteristics of autophagosomes are indicated by black arrows in PK-15 cell infected with PTV-2 (MOI = 0.1) at 9 h post-infection (hpi). Scale bar: 0.5 or 0.2 μm. (B) Formation of autophagosomes, shown as green punctae in PK-15/EGFP-LC3 cells that were infected with PTV-2 or treated with 2 μM rapamycin (Rapa) or left untreated (Mock). (C) Average number of punctae in each cell from at least 50 cells in each treatment. Data are reported as mean ± SEM of three independent experiments of panel B (n = 3; **P < 0.01). (D) PK-15/EGFP-LC3 cells were infected with PTV-2 for 9 h; the cells were then fixed, immunostained with anti-PTV-2 polyclonal antibody, and autophagosomes were observed by confocal microscopy. Scale bar: 10 μm. (E) and (F) Analysis of LC3-II expression in PK-15 cells infected with PTV-2 or mock-infected or treated with 2 μM rapamycin (Rapa), and its ratio to β-actin that was normalized to mock infection set at 1.0. Data are reported as mean ± SEM (n = 3; ns, not significant, *P < 0.05 and **P < 0.01)
PTV-2 infection induces autophagic responses without enhancing substrate degradation
Since some viruses induce the initial stage of autophagy but block autophagolysosomal fusion, we investigated whether PTV-2 induces a complete autophagic response. We first analyzed the p62 protein expression level. The p62 protein binds directly to LC3 and is degraded by autophagy. We did not observe obvious degradation of p62 during PTV-2 infection (Fig. 2A and B). Additionally, chloroquine (CQ, known to prevent autophagosome-lysosome fusion) was employed to analyze the turnover of autophagosomes. As shown in Fig. 2C and 2D, in mock-infected cells, levels of LC3-II increased markedly following treatment with CQ, indicating that autophagosome turnover and the degradation of LC3-II by lysosomal proteolysis was blocked. In contrast, in PTV-2-infected cells, no further accumulation of LC3-II was observed in response to CQ treatment, compared to the untreated controls, demonstrating that autophagosomes were not significantly turned over. This indicated that CQ had no observable effect on the degradation of LC3-II by lysosomal proteolysis in infected cells. We also determined p62 levels in CQ-treated PTV-2-infected cells. There was no significant increase (P > 0.05, Fig. 2C and 2E) in p62, revealing that degradation of autophagy substrates did not occur during PTV-2 infection.

Autophagosomes do not fuse with acidified proteolytic lysosomes in PTV-2-infected cells. (A) Western blotting analysis of p62 changes in PTV-2-infected PK-15 cells. (B) p62 to β-actin ratio normalized to mock infection (3 h) set at 1.0 of panel A (n = 3; *P < 0.05). (C), (D) and (E) PK-15 cells were mock-infected or infected with PTV-2 for 9 h. Subsequently, the cells were treated for 6 h in the absence or presence of the lysosomal inhibitor chloroquine (CQ), then LC3-II and p62 were analyzed by Western blotting and their ratios to β-actin were normalized to mock infection set at 1.0 (n = 3; ns, not significant and **P < 0.01). (F) Live cell imaging was performed to analyze colocalization of LysoTracker-stained acidified vesicles and EGFP-LC3-positive autophagosomes in uninfected and PTV-2-infected cells. PK-15/EGFP-LC3 cells were infected with PTV-2 for 9 h or left uninfected as control, then the cells were incubated with LysoTracker Red (50 nM) for 20 min. Scale bar: 10 μm. (G) Furthermore, fusion of autophagosomes with lysosomes was analyzed as colocalization of the autophagosome marker EGFP-LC3 with the lysosome marker LAMP1. PK-15/EGFP-LC3 cells were infected with PTV-2 for 9 h or left uninfected as a control, then the cells were incubated with lysosome marker LAMP1 (red) for 20 min. Scale bar: 10 μm
To further investigate why the accumulation of autophagosomes induced by PTV-2 did not lead to a higher protein degradation rate, we analyzed the level of autolysosomes in cells by staining stable EGFP-LC3 cells with LysoTracker, which labels and tracks acidic organelles such as lysosomes. PK-15/EGFP-LC3 cells were infected with PTV-2 for 9 h or left uninfected as a control. In PTV-2-infected cells, EGFP-LC3-positive vesicles did not localize with the LysoTracker staining (Fig. 2F). For further confirmation, we examined EGFP-LC3 colocalization with the lysosome-associated membrane protein 1 (LAMP1), a marker of endosomes and lysosomes. Co-localization was not observed (Fig. 2G). Taken together, our results suggest that PTV-2 infection induced an incomplete autophagy.
Cellular autophagy controls PTV-2 replication in cultured cells
Autophagy is involved in viral replication. To clarify the effects of autophagy on PTV-2 replication, we treated PK-15 cells with Wortmannin (WM) or 3-methyladenine (3-MA), class III phostphatidylinositol-3-kinase (PI3 K) inhibitors that are commonly used to suppress autophagy by blocking autophagosome formation, and investigated their effect on PTV-2 replication by measuring progeny virus yield. Both inhibitors reduced PTV-2-induced LC3-II (Fig. S3). Figure 3A shows that inhibition of autophagy by WM (P < 0.01) or 3-MA (P < 0.05) markedly increased the yield of PTV-2 progeny at 24 hpi. The results indicate that autophagy may control viral infection.

The effects of induction or inhibition of cellular autophagy on PTV-2 replication. (A) PTV-2 RNA copy numbers/ml determined by real time RT-PCR (left) and total progeny virus yield expressed as log TCID50/ml (right) in PTV-2-infected PK-15 cells treated with rapamycin (Rapa), Wortmannin (WM), 3-methyladenine (3-MA), siRNA targeting Atg5 (siAtg5), control scrambled siRNA (siCtrl) or left untreated (Ctrl) (n = 3; *P < 0.05 and **P < 0.01). (B) and (C) Effect of Atg5 silencing on LC3-II expression, and its ratio to β-actin
To make sure that these effects on PTV-2 were not due to the non-specific effects of these inhibitors, we suppressed the expression of Atg5, which is necessary for the activation of autophagosome formation, by RNA knockdown. PK-15 cells transfected with Atg5 siRNAs exhibited a significantly decreased level of endogenous Atg5 protein expression compared with cells transfected with non-targeting siRNAs (Fig. 3B and 3C, P < 0.01). The reduced Atg5 was accompanied by a higher yield of PTV-2 progeny (Fig. 3A, P < 0.05).
To further ascertain the effects of activated autophagy on PTV-2 replication, we induced cellular autophagy and examined its effect on PTV-2 progeny yield. In contrast, induction of autophagy following rapamycin treatment decreased the yield of PTV-2 progeny (Fig. 3A, P < 0.05). Together, these findings suggest that cellular autophagy is involved in regulating PTV-2 replication, although we cannot rule out other effects that these chemical treatments might exert on the virus life cycle, independent of autophagy.
Discussion
Autophagy is an organized and destructive mechanism that degrades and recycles long-lived proteins or dysfunctional cellular components [14]. Previous studies suggest that autophagy plays an important part in the antiviral defense mechanism; however, the role of autophagy in the viral life cycle and pathogenesis is complicated and may have different outcomes. Subversion of the autophagic pathway also allows pathogens to establish a replicative niche and supplies nutrients for growth [15].
The manipulation of autophagy has been confirmed in some RNA viruses including Newcastle disease virus (NDV) [16,17,18], dengue virus (DENV) [19], Japanese encephalitis virus (JEV) [20, 21], and hepatitis C virus (HCV) [22, 23]. Autophagy may also be a mechanism of eliminating intracellular viruses, such as herpes simplex virus (HSV-1) [24] and tobacco mosaic virus [25]. Viruses classified within the family Picornaviridae are well-known for manipulating the autophagy pathway and exploiting it to enhance replication [10], since they rely heavily on cellular membranes at numerous stages of their infectious cycle [26]. The entire replication cycle of picornaviruses takes place in the cytosol [27]. Therefore, by inducing autophagy, these viruses may facilitate the creation of scaffolds for their own replication. Enterovirus 71(EV71)-induced autophagy promotes viral replication in vitro and in vivo [28]. While poliovirus and coxsackie virus B3 (CVB3) both subvert autophagy, the two viruses have very different effects on the autophagic pathway [11]. Foot-and-mouth-disease virus (FMDV), perhaps the best-known non-human picornavirus, utilizes an autophagic pathway to favor its own replication [29]. Another study demonstrated that FMDV induces autophagosomes during cell entry to facilitate infection, but not to provide membranes for replication [30]. In a report by Zhang et al. [31], it was suggested that autophagy promotes encephalomyocarditis virus (EMCV) replication and autophagosomes mediate non-lytic release of cytoplasmic viruses. The finding, together with other studies using enterovirus and poliovirus, support the hypothesis that autophagy-derived membranes rather serve as means of non-lytic virus spread than as a scaffold for viral genome replication [32]. Human rhinovirus 1A (HRV-1A), however, did not induce or subvert autophagy [33], indicating that there is virus to virus variation in the membrane generating pathways and not all viruses use the autophagic machinery in the same way. However, until now it has remained unclear whether autophagy is involved in PTV-2 infection, and if so, what role it plays in virus replication.
Our results showed that PTV-2 induces the accumulation of autophagosomes in PK-15 cells (Fig. 1A). Increased amounts of EGFP-LC3 punctae (Fig. 1B and 1C) were observed in PTV-2-infected cells (Fig. 1D). We also detected colocalization of viral proteins and the autophagy marker LC3. Previous studies investigating picornavirus infections have demonstrated that viral replicase proteins colocalized with LC3 during infection (e.g. poliovirus and CVB3) [34, 35], indicating that the viruses may utilize the autophagic membranes for replication.
The autophagosome fuses with lysosomes to generate autolysosomes, which are recycled or in the case of infection, provide antigens for presentation via the MHC molecules [36]. Interestingly, during PTV-2 infection, autophagic flux was not enhanced. This lack of enhancement of autophagic flux is measured by p62 degradation and autophagosome turnover. p62 can bind LC3, serving as a selective substrate of autophagy [37]. The protein level of p62 was not affected during virus infection as compared to uninfected controls (Fig. 2A and 2B). In addition, the endogenous LC3-II accumulated significantly in the presence of CQ in mock-infected cells. However, no differences in LC3-II or p62 levels were observed in PTV-2-infected cells treated with CQ, when compared to the untreated controls (Fig. 2C, 2D and 2E), indicating that PTV-2 infection may play a similar role to CQ in blocking autophagosome maturation. For further confirmation, the autophagosome-lysosome fusion process was measured, and we observed inefficient fusion between autophagosomes and lysosomes (Fig. 2F and 2G). This suggests that PTV-2 may prevent the fusion of the lysosomes and autophagosomes and thus inhibit the degradation process. One plausible assumption is that viruses have evolved strategies to antagonize this process. For example, HSV-1 neurovirulence protein ICP34.5 or viral Bcl-2 s encoded by the γ-herpesviruses interact with Beclin1, thereby inhibiting autophagy and facilitating viral replication [38]. A similar strategy is also seen during FMDV infection; binding of 2C to Beclin1 blocks the fusion of FMDV-containing autophagosomes to lysosomes, preventing virus degradation [39].
The autophagy pathway is now playing a central role in the immunological control of viral infection. As an adversarial counterpoint, viruses have evolved to block the autophagic attack and deploy their own autophagic machinery for survival [40]. Knockdown of autophagy-related genes has been shown to be detrimental to viral yields, e.g. for the mammalian picornavirus poliovirus [41], a phenomenon that might also apply for other picornaviruses. Intriguingly, a Drosophila melanogaster picornavirus also replicates in association with double-membrane vacuoles but autophagy genes are not required for its replication [42]. This suggests that the requirement for the autophagic machinery in the generation of double-membrane vacuoles that support picornavirus replication may be cell-type specific or restricted to certain hosts [43]. Therefore, we sought to determine whether autophagy is a weapon against PTV-2 infection or a process hijacked by the virus or dispensable for viral replication. Cells treated with the autophagy inhibitors WM or 3-MA, which obstruct autophagic processes, elevated virus titer. Correspondingly, autophagy inhibition, through Atg5 knockdown, led to increases in virus yield. In contrast, induction of autophagy by rapamycin lowered PTV-2 titers (Fig. 3A). These results indicate that activation of cellular autophagy is a defense mechanisms targeting PTV-2. Similarly, depletion of Atg5 impairs central nervous system clearance of Sindbis virus (SINV) capsid and increases the mortality of infected mice [8]. The antiviral function of autophagy indicates that therapeutic autophagy upregulation may present a promising strategy for treating viral infections. For instance, autophagy induction by vitamin D or rapamycin has been shown to hinder HIV replication in primary human macrophages in vitro [44]. In addition, a cell-permeable autophagy-inducing peptide (Tat-beclin 1) restricts the replication of several viruses in vitro, including HIV-1, SINV, chikungunya virus (CHIKV), and West Nile virus (WNV), and also reduces the mortality of neonatal mice infected with either CHIKV or WNV [45]. Therefore, exploring therapeutic strategies, by modulating specific stages of autophagy, may help controlling viral diseases.
This report constitutes the first demonstration that PTV-2 induces the accumulation of autophagosomes, and this process seems to hinder PTV-2 replication. However, PTV-2 does not enhance autophagic protein degradation. Our study provides insights into PTV-2-host interactions and increases our knowledge of the roles of autophagy in the virus life cycle, although deciphering the mechanism involved needs further research, e.g. the origin of the double-membraned vesicles and the viral protein(s) involved in autophagy induction. Such work may lead to a better understanding of PTV-2 pathogenesis.
References
Knowles NJ, Hovi T, Hyypiä T (2012) Family Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Classification and nomenclature of viruses: ninth report of the international committee on taxonomy of viruses. Elsevier, San Diego, pp 855–880
Chiu SC, Hu SC, Chang CC, Chang CY, Huang CC, Pang VF, Wang FI (2012) The role of porcine teschovirus in causing diseases in endemically infected pigs. Vet Microbiol 161:88–95
Tsai ATH, Kuo CC, Kuo YC, Yang JL, Chang CY, Wang FI (2016) The urinary shedding of porcine teschovirus in endemic field situations. Vet Microbiol 182:150–155
Chiu SC, Yang CL, Chen YM, Hu SC, Chiu KC, Lin YC, Chang CY, Wang FI (2014) Multiple models of porcine teschovirus pathogenesis in endemically infected pigs. Vet Microbiol 168:69–77
Jones TH, Muehlhauser V (2017) F-coliphages, porcine adenovirus and porcine teschovirus as potential indicator viruses of fecal contamination for pork carcass processing. Int J Food Microbiol 241:237–243
Wang B, Tian Z-J, Gong D-Q, Li D-Y, Wang Y, Chen J-Z, An T-Q, Peng J-M, Tong G-Z (2010) Isolation of serotype 2 porcine teschovirus in China: evidence of natural recombination. Vet Microbiol 146:138–143
Jackson WT (2015) Viruses and the autophagy pathway. Virology 479:450–456
Orvedahl A, MacPherson S, Sumpter R, Tallóczy Z, Zou Z, Levine B (2010) Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127
Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV-1 ICP34. 5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35
McKnight KL, Lemon SM (2017) Virology: Ins and outs of picornaviruses. Nature 541:299–300
Lai JK, Sam I, Chan YF (2016) The autophagic machinery in enterovirus infection. Viruses 8:32
Zhu B, Xu F, Li J, Shuai J, Li X, Fang W (2012) Porcine circovirus type 2 explores the autophagic machinery for replication in PK-15 cells. Virus Res 163:476–485
Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y, Ye C, Liao M, Zhou J (2015) Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 11:503–515
Levine B, Klionsky DJ (2017) Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci USA 114:201–205
Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T (2015) Autophagy and autophagy-related proteins in the immune system. Nat Immunol 16:1014
Kang Y, Yuan R, Xiang B, Zhao X, Gao P, Dai X, Liao M, Ren T (2017) Newcastle disease virus-induced autophagy mediates antiapoptotic signaling responses in vitro and in vivo. Oncotarget. https://doi.org/10.18632/oncotarget.18169
Cheng J-H, Sun Y-J, Zhang F-Q, Zhang X-R, Qiu X-S, Yu L-P, Wu Y-T, Ding C (2016) Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci Rep 6:24721
Sun Y, Yu S, Ding N, Meng C, Meng S, Zhang S, Zhan Y, Qiu X, Tan L, Chen H, Song C, Ding C (2014) Autophagy Benefits the Replication of Newcastle Disease Virus in Chicken Cells and Tissues. J Virol 88:525–537
Green AM, Beatty PR, Hadjilaou A, Harris E (2014) Innate immunity to dengue virus infection and subversion of antiviral responses. J Mol Biol 426:1148–1160
Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G (2013) Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One 8:e52909
Li J-K, Liang J-J, Liao C-L, Lin Y-L (2012) Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 14:159–168
Chan ST, Lee J, Narula M, Ou J-HJ (2016) Suppression of Host Innate Immune Response by Hepatitis C Virus via Induction of Autophagic Degradation of TRAF6. J Virol 90:10928–10935
Dreux M, Chisari F (2009) Autophagy proteins promote hepalitis C virus replication. Autophagy 5:1224–1225
Tallóczy Z, Virgin I, Herbert Levine B (2006) PKR-dependent xenophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29
Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar S (2005) Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577
Mateo R, Nagamine CM, Spagnolo J, Méndez E, Rahe M, Gale M, Yuan J, Kirkegaard K (2013) Inhibition of cellular autophagy deranges dengue virion maturation. J Virol 87:1312–1321
Richards AL, Jackson WT (2013) Behind closed membranes: the secret lives of picornaviruses? PLoS Pathog 9:e1003262
Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS (2009) Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol 81:1241–1252
O’donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, Borca MV, Baxt B (2011) Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 410:142–150
Berryman S, Brooks E, Burman A, Hawes P, Roberts R, Netherton C, Monaghan P, Whelband M, Cottam E, Elazar Z (2012) Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J Virol 86:12940–12953
Zhang Y, Li Z, Ge X, Guo X, Yang H (2011) Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 7:613–628
Bird SW, Maynard ND, Covert MW, Kirkegaard K (2014) Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci USA 111:13081–13086
Quiner CA, Jackson WT (2010) Fragmentation of the Golgi apparatus provides replication membranes for human rhinovirus 1A. Virology 407:185–195
Delorme-Axford E, Morosky S, Bomberger J, Stolz DB, Jackson WT, Coyne CB (2014) BPIFB3 regulates autophagy and coxsackievirus B replication through a noncanonical pathway independent of the core initiation machinery. MBio 5:e02114–e02147
Taylor MP, Kirkegaard K (2008) Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 4:286–289
Münz C (2016) Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol 37:755–763
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145
O’Connell D, Liang C (2016) Autophagy interaction with herpes simplex virus type-1 infection. Autophagy 12:451–459
Gladue D, O’donnell V, Baker-Branstetter R, Holinka L, Pacheco J, Fernandez-Sainz I, Lu Z, Brocchi E, Baxt B, Piccone M (2012) Foot-and-mouth disease virus nonstructural protein 2C interacts with Beclin1, modulating virus replication. J Virol 86:12080–12090
Dong X, Levine B (2013) Autophagy and viruses: adversaries or allies? J Innate Immun 5:480–493
Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3:e156
Cherry S, Kunte A, Wang H, Coyne C, Rawson RB, Perrimon N (2006) COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog 2:e102
Levine B, Deretic V (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 7:767–777
Nardacci R, Ciccosanti F, Marsella C, Ippolito G, Piacentini M, Fimia GM (2017) Role of autophagy in HIV infection and pathogenesis. J Intern Med 281:422–432
Shoji-Kawata S, Sumpter R Jr, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D (2013) Identification of a candidate therapeutic autophagy–inducing peptide. Nature 494:201
Acknowledgements
This work is partly supported by the important agriculture subject fund from Department of S&T of Zhejiang Province (2015C02044), Department of Education of Zhejiang Province (Y201635576), the Agricultural Technology Extension Funds of Zhejiang University, Dabei Agricultural Discipline Development and Talent Training Fund (2017ZDNT004), and three rural six party funds for Xiaoliang Li.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
All animal studies were approved by the Animal Care and Use Committee of Zhejiang University in accordance with the Chinese guidelines for the care and use of laboratory animals (Permit Number: 2016101098).
Additional information
Handling Editor Zhenhai Chen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gu, Y., Zhou, Y., Shi, X. et al. Porcine teschovirus 2 induces an incomplete autophagic response in PK-15 cells. Arch Virol 163, 623–632 (2018). https://doi.org/10.1007/s00705-017-3652-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3652-2