
Abstract
Common reed (Phragmites australis) plants showing chlorotic stripe symptoms on leaves were found in Gansu Province, China. Deep sequencing of small RNAs from symptomatic leaves identified a putative potyvirus, which was named common reed chlorotic stripe virus (CRCSV). The full genome sequence was determined by reverse transcription PCR, rapid amplification of cDNA ends (RACE) PCR, and sequencing. It consists of 9,426 nucleotides, excluding the poly(A) tail, and contains a large open reading frame encoding a polyprotein of 3,014 amino acids. Putative proteolytic cleavage sites were identified. Since CRCSV shared low sequence similarity (35%-37% identity) to any known members of the family Potyviridae and it clustered uniquely in phylogenetic analysis of either the polyprotein or the coat protein, CRCSV is a distinct, previously undescribed member of the family Potyviridae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The Potyviridae are one of the largest and most economically important plant virus families, consisting of eight genera based on genome organization, vector transmission, genetic relatedness, host range, key host reactions, different inclusion body morphology and antigenic properties [9, 11]. Members of this family have a genome of positive-sense RNA, approximately 10 kb long, with a virus-encoded protein (VPg) covalently attached to the 5′ terminus, and the 3′ terminus is polyadenylated [8, 15]. There is a single long open reading frame (ORF) encoding a polyprotein that is post-translationally processed into the individual gene products by viral proteases [15]. Additionally, a small ORF, pipo, overlaps the P3 region of the polyprotein ORF in the −1/+2 reading frame [4]. Recently, another small ORF, pispo, which overlaps the P1 coding region in the −1/+2 frame was discovered in members of the sweet potato feathery mottle virus (SPFMV) subgroup [14].
Common reed (Phragmites australis (Cav.) Trin. ex Steu) is a perennial weed in the family Poaceae with wide adaptability, strong resistance, and rich germplasm resources that spreads rapidly by rhizome and seed in China [12]. It has become an economically important resource plant in northwestern China as a raw material for paper making, excellent forage, and for the redevelopment of contaminated land and maintaining ecological balance [10]. It has been reported to be a reservoir host of barley yellow dwarf virus-PAV, maize dwarf mosaic virus and sugarcane mosaic virus in Turkey, resulting in mosaic, chlorosis and streak symptoms [7]. However, to our knowledge, no novel virus has been directly identified in common reed.
In July 2015, common reed plants showing virus disease symptoms such as systemically streaked chlorosis and necrosis on leaves (Fig. 1A) were observed in Tianshui city, Gansu province, China. Symptomatic leaf samples were collected, and total RNA was isolated using TRIzol Reagent (Tiangen, Beijing, China). A small RNA library was prepared using 2.0 μg of total RNA and a mirVana™ miRNA Isolation Kit using the standard protocol (Austin TX, USA) and sequenced in a single lane on a HiSeq 2500 platform (Illumina). The next-generation sequencing (NGS)-generated reads were trimmed to remove low-quality and adapter sequences and then assembled de novo into larger contigs using the ABySS Software 1.9.0 (http://www.bcgsc.ca/platform/bioinfo/software/abyss) with a k-mer of 25. The assembled contigs were further analyzed, using local BLASTn search programs, against the viral sequences in the GenBank Virus Reference Database (http://www.ncbi.nlm.nih.gov). Based on the BLASTn search results, only three contigs (2536 bp, 2749 bp and 4076 bp in length) were found to share high levels of sequence identity with several potyviruses, i.e., the first contig shared 67.3% sequence identity with tomato necrotic stunt virus (GenBank accession number JQ314463.1), the second shared 66.7% identity with verbena virus Y (EU564817.1), and the third shared 64.6% identity with chilli ringspot virus (KM229702). We therefore concluded that the virus infecting common reed plant might be a potyvirus.

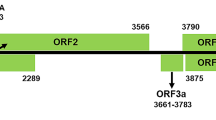
Characterization of common reed chlorotic stripe virus (CRCSV), a new member of the family Potyviridae. (A) Virus disease symptoms on common reed plants found in Gansu province, China. (B) Schematic representation of the genomic organization of CRCSV. The large open reading frame (ORF) is depicted by an open box. Numbers above the diagram represent the first nucleotide of each cistron. The position of the gene encoding the putative protein PIPO is also shown. Putative amino acid cleavage sites are indicated at the bottom of the diagram. (C and D) Phylogenetic analysis of the polyprotein amino acid sequences (C) and the coat protein amino acid sequences (D) of CRCSV and other selected members of different genera in the family Potyviridae using the neighbor-joining algorithm. Bootstrap analysis was applied using 1000 replicates. The scale bar represents a genetic distance of 0.2. The following viruses were included in the analysis: scallion mosaic virus (ScaMV, CAC87085.1), lupine mosaic virus (LuMV, ACJ31798.2), papaya ringspot virus (PRSV, AAG47346.1), sugarcane mosaic virus (SCMV, AMM72620.1), potato virus Y (PVY, AKG94974.1), tobacco vein mottling virus (TVMV, CAA27720.1), johnsongrass mosaic virus (JGMV, ALS88434.1), pepper mottle virus (PepMoV, AAA47910.1), ryegrass mosaic virus (RGMV, AAC25028.1), agropyron mosaic virus (AgMV, AAS77619.2), hordeum mosaic virus (HoMV, AAS65455.2), barley mild mosaic virus (BaMMV, AAQ10758.1), barley yellow mosaic virus (BaYMV, CAA10637.1), oat mosaic virus (OMV, CAC84680.1), wheat yellow mosaic virus (WYMV, BAA28768.1), sweet potato mild mottle virus (SPMMV, CAA97466.1), cassava brown streak virus (CBSV, ADR73022.1), squash vein yellowing virus (SqVYV, ABY86626.1), cucumber vein yellowing virus (CVYV, AAT66639.1), wheat streak mosaic virus (WSMV, AAC13692.1), brome streak mosaic virus (BrSMV, CAA88417.1), oat necrotic mottle virus (ONMV, AAQ91884.1), blackberry virus Y (BVY, AAX87001.1), Chinese yam necrotic mosaic virus (CYNMV, BAM36463.1), caladenia virus A (CalVA, AFQ95549.1), triticum mosaic virus (TriMV, ABO41208.2), and sugarcane streak mosaic virus (SCSMV, ADE34528.1)
To confirm the NGS results, first-strand cDNA was synthesized by reverse transcription of total RNA isolated from reed samples with M-MLV reverse transcriptase (Promega) and an oligo (dT) primer and subjected to PCR using several pairs of primers designed from the NGS contig sequences (Supplementary Table S1). The above three contigs (2536 bp, 2749 bp and 4076 bp in length) were confirmed and reconstructed by PCR and sequencing. The internal gaps between the three contigs were amplified by RT-PCR with specific primers (Supplementary Table S1). Analysis of the larger assembled sequence using DNAMAN6.0 software indicated that it contained a large complete ORF (Fig. 1B). To complete the full nucleotide sequence of this new virus, the 5′-UTR and 3′-UTR sequences were determined by rapid amplification of cDNA ends (RACE) PCR. The full genomic sequence of this new virus was 9, 426 nucleotides long. BLASTx search with the complete nucleotide sequence revealed that it shared 35%-37% amino acid sequence identity with several potyviruses, including turnip mosaic virus, jasmine virus T, ornithogalum mosaic virus, sunflower mild mosaic virus and Japanese yam mosaic virus, with 88%-93% query coverage. This new virus was tentatively named common reed chlorotic stripe virus (CRCSV). The genome sequence has now been deposited in the GenBank database under the accession number KY612317.
The CRCSV genome encodes a single large ORF 9204 nt (3067 amino acids) in length starting at the first AUG (nt 186-188) and ending at the ochre stop codon UAA (nt 9387-9389), encoding a polyprotein of 339.1 kDa (Fig. 1B). BLASTx results indicated that nine putative cleavage sites were predicted at amino acid positions 239, 697, 1043, 1096, 1735, 1788, 2005, 2245 and 2758, and thus the polyprotein is predicted to be proteolytically processed into ten mature proteins P1, HC-Pro, P3, 6K1, CI, 6K2, NIa-VPg, NIa-Pro, NIb and CP (Fig. 1B) [2]. Putative cleavage sites were compared, and the results indicated that HC-Pro/P3 of CRCSV is highly conserved compared with other members belonging to the different genera in the family Potyviridae, while the other cleavage sites show diversity and flexibility (Supplementary Table S2). The small ORF pipo was also predicted starting from a G2A6 motif at position 2722. This motif is similar to the highly conserved G1-2A6-7 motif that is present in other members in the family Potyviridae [4]. Analysis of typical conserved motifs indicated that the serine protease catalytic residues 163H-8X-D-33X-S206 (X, any amino acid residue) are conserved in the C-terminal part of the P1 protein [5]. The FRNK motif in HC-Pro, which is associated with symptom expression [6], was changed to FRNT428, and the KITC and PTK motifs, which are tightly associated with potyvirus transmission by aphids, were changed to LFQC299 and PIE548, respectively [15]. G-A-V-G-S-G-K-S-T and V-LL-I-E-P-T-R-P-L motifs in the CI protein were changed to 1185G-P-V-G-S-G-K-S-T1193 and 1205V-L-V-L-E-P-T-R-P-L1214 [5]. In addition, the conserved motif 2056H-34X-D-67X-G-X-C-G-14X-H2176 was found in NIa-Pro [5]. The C-D-A-D-G-S motif of NIb was changed to 2492C-H-A-D-G-S2497, while the 2557S-G-3X-T-3X-N-T-30X-G-D-D2596 was conserved [15]. The D-A-G motif in the N-terminus of the CP of most potyviruses, which is important for aphid transmission [3], was changed to 2765D-A-E2767 in CRCSV. All these motif changes were confirmed by RT-PCR and sequencing.
We attempted to perform CRCSV transmission experiments on a range of indicator plants via rub inoculation. Crude sap was prepared from symptomatic young leaves in 0.1 M phosphate buffer (pH 7.0) in a ratio of 1: 5 (w/v). Seven species of indicator plants of the families Caricaceae, Chenopodiaceae and Solanaceae were inoculated (Supplementary Table S3). Infection phenotype was monitored every two days post-inoculation for one month. No local lesions were observed on any inoculated leaves. Cucumber plants showed slight chlorosis symptoms on upper leaves at 20 days post-inoculation (Supplementary Fig. S1A), while other species showed no symptoms. RT-PCR was performed to detect CRCSV in upper leaves of all inoculated plants, using two primers (CRCSV CP-2F and CP-2R) specific for the CRCSV cp gene sequence (Supplementary Table S1). The results showed that CRCSV could be detected in nine of ten inoculated cucumber seedlings (Supplementary Fig. S1B). The result was confirmed by cloning and DNA sequencing.
To determine the taxonomic position of CRCSV, phylogenetic trees based on the polyprotein and CP sequences of CRCSV and 27 other members in the family Potyviridae representing all eight genera were constructed using the neighbor-joining method implemented in the MEGA6.06 program using sequence alignments generated by Clustal X [13]. Bootstrap analysis was applied using 1000 replicates. In both trees, CRCSV was most closely related to blackberry virus Y (BVY, the only member of the genus Brambyvirus) (Fig. 1C and D), but it failed to cluster with any members other genera, suggesting that CRCSV might represent a new genus within the family Potyviridae.
References
Adams MJ, Antoniw JF, Fauquet CM (2005) Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol 150:459–479
Adams MJ, Antoniw JF, Beaudoin F (2005) Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol Plant Pathol 6:471–487
Atreya CD, Raccah B, Pirone TP (1990) A point mutation in the coat protein abolishes aphid transmissibility of a potyvirus. Virology 178:161–165
Chung BY, Miller WA, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA 105:5897–5902
Deng CL, Wang WJ, Wang ZY, Jiang X, Cao YY, Zhou T, Fan ZF (2008) The genomic sequence and biological properties of Pennisetum mosaic virus, a novel monocot-infecting potyvirus. Arch Virol 153:921–927
Gal-On A (2000) A point mutation in the FRNK motif of the potyvirus helper component-protease gene alters symptom expression in cucurbits and elicits protection against the severe homologous virus. Phytopathology 90:467–473
Ilbağı H (2006) Common reed (Phragmites communis) is a natural host of important cereal viruses in the Trakya region of Turkey. Phytoparasitica 34:441–448
Ivanov KI, Eskelin K, Lõhmus A, Mäkinen K (2014) Molecular and cellular mechanisms underlying potyvirus infection. J Gen Virol 95:1415–1429
King AMQ, Lefkowitz E, Adams MJ, Castens EB (eds) (2012) Virus taxonomy: ninth report of the international committee on taxonomy of viruses, vol 9. Academic Press, London
Mal TK, Narine L (2004) The biology of Canadian weeds. 129. Phragmites australis (Cav.) Trin. ex Steud. Can J Plant Sci 84:365–396
Revers F, Le Gall O, Candresse T, Maule AJ (1999) New advances in understanding the molecular biology of plant/potyvirus interactions. Mol Plant Microbe Interact 12:367–376
Schröder P, Daubner D, Maier H, Neustifter J, Debus R (2008) Phytoremediation of organic xenobiotics–Glutathione dependent detoxification in Phragmites plants from European treatment sites. Bioresource Technol 99:7183–7191
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Untiveros M, Olspert A, Artola K, Firth AE, Kreuze JF, Valkonen J (2016) A novel sweet potato potyvirus open reading frame (ORF) is expressed via polymerase slippage and suppresses RNA silencing. Mol Plant Pathol 17:1111–1123
Urcuqui-Inchima S, Haenni AL, Bernardi F (2001) Potyvirus proteins: a wealth of functions. Virus Res 74:157–175
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT1042).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This study did not include experiments with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yuan, W., Du, K., Fan, Z. et al. Complete genomic sequence of common reed chlorotic stripe virus, a novel member of the family Potyviridae . Arch Virol 162, 3541–3544 (2017). https://doi.org/10.1007/s00705-017-3454-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3454-6