
Abstract
Apple stem grooving virus (ASGV), apple chlorotic leaf spot virus (ACLSV), and prunus necrotic ringspot virus (PNRSV) were identified in a crab apple tree by small RNA deep sequencing. The complete genome sequence of ACLSV isolate BJ (ACLSV-BJ) was 7554 nucleotides and shared 67.0%-83.0% nucleotide sequence identity with other ACLSV isolates. A phylogenetic tree based on the complete genome sequence of all available ACLSV isolates showed that ACLSV-BJ clustered with the isolates SY01 from hawthorn, MO5 from apple, and JB, KMS and YH from pear. The complete nucleotide sequence of ASGV-BJ was 6509 nucleotides (nt) long and shared 78.2%-80.7% nucleotide sequence identity with other isolates. ASGV-BJ and the isolate ASGV_kfp clustered together in the phylogenetic tree as an independent clade. Recombination analysis showed that isolate ASGV-BJ was a naturally occurring recombinant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Crab apples belong to the genus Malus in the family Rosaceae and are popular ornamental trees in the commercial and residential landscape for their beautiful flowers or fruit [6]. They exhibit excellent ornamental characteristics and stress resistance and can thus be used as rootstocks for apples and potential pollinizers in commercial orchards. Crab apples are subject to the same pathogens as apples, including several viruses such as apple chlorotic leaf spot virus (ACLSV), apple stem grooving virus (ASGV) and apple stem pitting virus (ASPV). There are several viruses or virus strains that are latent in apple varieties but which can cause serious alterations in the leaves, fruit, and sterns of some crab apple varieties that can be used as indicators for detection of latent virus infections of apples [8].
Over the last decade, advances in DNA sequencing technology have led to the development of new approaches for the identification and detection of viruses and viroids [1, 20]. These new approaches involve sequencing of the total nucleic acid or small RNAs in samples from diseased plants by next-generation sequencing (NGS) and allow identification of pathogens by downstream analysis using bioinformatic tools. In the past few years, NGS using various technologies and templates (RNAs, DNA, short interfering RNAs, double-stranded RNAs) has contributed tremendously to unraveling the nature of microbial agents associated with disease and allowed the rapid identification and characterization of a number of novel or known plant viruses with an RNA or DNA genome [2, 3, 10].
Ornamental crab apples showing virus-like symptoms such as necrosis and chlorosis of leaves and decline of tree vigor were observed in a landscaped park in Beijing. To identify the potential viruses involved in the disease, symptomatic leaves were collected, and total RNA was extracted from the samples using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Total RNA was then quantified and assessed for quality using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE) and integrity using a Bio-Analyzer 2100 (Agilent Technologies, Waldbronn, Germany) and used for construction of a small RNA library with subsequent sequencing on a HiSeq2000 platform (Illumina). Sequence reads were trimmed to remove low-quality and adaptor sequences and assembled using the de novo assembly algorithm of Velvet [22]. The assembled contigs were analyzed by BLASTn and BLASTx searches against the GenBank database.
A total of 23,140,206 high-quality reads were obtained using the Illumina Hiseq 2000 Solexa platform after quality trimming, 12,700,358 of which were virus-derived. 5727 contigs were assembled using the Velvet program with a k-mer value of 17, with the size ranging from 33 to 364 nt. A BLASTn and BLASTx analysis of assembled contigs against the NCBI database, using a high-homology sequence search, identified 79 contigs showing high sequence similarity to ASGV (61 contigs, with 46% sequence coverage), ACLSV (nine contigs, with 7% sequence coverage) and PNRSV (nine contigs, with 7% sequence coverage), while all the remaining contigs were of host origin. RT-PCR was conducted with primers designed based on the assembled contigs, and this confirmed the presence of these three viruses. PNRSV, a member of the genus Ilarvirus in the family Bromoviridae, has been documented in peach, apple, plum, cherry and apricot in China [5] and was only partially sequenced in this study (GenBank accession number KX371574). BLAST analysis showed that this sequence shared 75% nucleotide sequence identity (the highest) with the corresponding region of the isolate ChrYL (GenBank accession number KT444702).
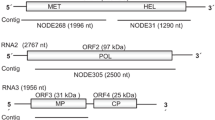
The complete genome sequence of ACLSV-BJ was obtained by Sanger sequencing RT-PCR and rapid amplification of cDNA ends (RACE) PCR for the 5′ and 3′ ends of the virus genome (Supplemental Table 1) and submitted to the GenBank database with the accession number KU960942. The genomic RNA of ACLSV-BJ was 7554 nucleotides in length excluding the 3′ polyA tail, and the 5′ and 3′ ends were 154 and 210 nt, respectively. Sequence analysis showed that the genome nucleotide sequence identities between ACLSV-BJ and other ACLSV isolates available in the GenBank database ranged from 67.0% (TaTao5) to 83.0% (SY01). The genomic organization of ACLSV-BJ was the same as that of previously described ACLSV isolates and consisted of three ORFs. ORF 1 (nt 155-5788) putatively encoded a 1877-aa long viral replicase polyprotein sharing 91% amino acid sequence identity with the Chinese pear isolates KMS, JB and YH [23] and recently described hawthorn isolate SY01 [18]. ORF2 (nt 5700-6983) encoded a 47-kDa movement protein and shared 93% amino acid sequence identity with the isolates KMS, JB, YH and SY01. BLAST analysis showed that the coat protein encoded by ORF3 (nt 6766-7344) shared 89-96% aa sequence identity with other isolates. A previous analysis based on the amino acid sequences of CP showed that the ACLSV isolates were separated into two major clusters in which the combinations of the five amino acids at positions 40, 59, 75, 130 and 184 (S40-L59-Y75-T130-L184or A40-V59-F75-S130-M184) were highly conserved within each cluster [21]. In the CP of ACLSV-BJ, the motif S40-M59-Y75-A130-S184 was identified. Moreover, the specific aa combination S73-D82-L83-G98 reported for isolates JB, KMS, YH, KRL (a kuerle isolate) and MO5, which clustered together in the phylogenetic tree, was also conserved in the CP of ACLSV-BJ (S71-D81-L82-G97). A phylogenetic tree (Fig. 1A) based on the complete genome sequences of ACLSV isolates aligned using Clustal W [17] was constructed using the neighbor-joining (NJ) method implemented in the MEGA6.06 program [17] with the best-fit model (Tamura-Nei model) recommended by a model test. This analysis showed that ACLSV-BJ clustered with the isolates SY01 from hawthorn, MO5 from apple, and JB, KMS and YH from pear, which was in accordance with the nt and aa sequence analyses.

Neighbor-joining tree generated based on the complete genome nucleotide sequences of isolates of apple chlorotic leaf spot virus (A) and apple stem grooving virus (B). Data on geographic provenance and hosts are shown next to the GenBank accession number for each isolate used in the analysis. The number of bootstrap replicates was 1000. Branches with bootstrap values ≥70% are shown. The scale bar represents genetic distance (substitutions per nucleotide)
The complete genome sequence of ASGV-BJ was also determined in this work and deposited in GenBank under the accession number KU947036. The complete nucleotide sequence of ASGV-BJ was 6509 nucleotides (nt) long, excluding the poly (A) tail. The genomic RNA had two overlapping ORFs. ORF1 (nt 42–6351) encoded a 2,105-amino-acid (aa) polypeptide containing methyltransferase-like, papain-like protease, helicase-like, and RdRp-like domains, and the coat protein located at the carboxy-terminal end of the polyprotein. This ORF was preceded by a 41-nucleotide-long non-coding region at the 5′ end of the genome and followed by a 150-nucleotide-long intergenic region. ORF2 (nt 4793-5755) encoded a 320-amino-acid polypeptide, a protein with conserved motifs for both movement proteins (MPs), and viral proteases. Sequence analysis showed that ASGV-BJ shared 78.2-80.7% nucleotide sequence identity with other isolates. In a previous study, the transcription start sites of both the CP and the movement protein (MP) sgRNAs of ASGV were mapped to a conserved hexanucleotide motif, UUAGGU, upstream of each sgRNA [9]. This conserved hexanucleotide motif was also found at nt 4647, upstream of the movement protein gene, and similarly, at nt 5607, upstream of the CP gene in the ASGV-BJ isolate. To elucidate the phylogenetic relationship of ASGV-BJ to other isolates, a phylogenetic tree was constructed using the available complete ASGV genome sequences. ASGV-BJ clustered together with the isolate ASGV_kfp (KR106996) from pear (Fig. 1B).
Recombination plays a significant role in the evolution of many viruses [13, 14, 19] and is well documented among plant-infecting single-stranded RNA viruses [4, 15]. Recombination analysis of ASGV isolates was carried out using the software RDP3 [11], which includes RDP, GENECONV, BOOTSCAN, MAXCHI, CHIMAERA, SISCAN, and 3SEQ, performed with the default configuration, except that the options of linear sequence and of disentangling overlapping signals were selected. An event detected by at least five different methods and with p-values < 10−6 was considered to be a positive recombination event. Of the 20 genomes analyzed, ten of them showed evidence of recombination (Table 1), indicating a relatively high recombination frequency within the population of fully sequenced ASGV isolates. Crossover sites were identified at different locations for these recombinants, suggesting that there were no hotspots. ASGV-BJ was found to be a recombinant originating from isolate ASGV_kfp (KR106996) and an isolate from South Korea associated with pear black necrotic leaf spot disease (AY596172). Recombination in ACLSV has been reported previously [7]. Our analyses confirmed previous reports, with no additional events detected in this study.
References
Adams IP, Glover RH, Monger WA, Mumford R, Jackeviciene E, Navalinskiene M, SamuitieneM Boonham N (2009) Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol. Plant Pathol. 10:537–545
Al Rwahnih M, Daubert S, Golino D, Rowhani A (2009) Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 387:395–401
Al Rwahnih M, Dave A, Anderson MM, Rowhani A, Uyemoto JK, Sudarshana MR (2013) Associationof a DNA virus with grapevines affected by red blotch disease in California. Phytopathology 103:1069–1076
Chare ER, Gould EA, Holmes EC (2003) Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 84:2691–2703
Cui HG, Liu HZ, Chen J, Zhou JF, Qu LN, Su JM, Wang GP, Hong N (2015) Genetic diversity of Prunus necrotic ringspot virus infecting stone fruit trees grown at seven regions in China and differentiation of three phylogroups by multiplex RT-PCR. Crop Prot. 74:30–36
den Boer AF (1959) Ornamental crab apples. Am Assoc Nurserymen, Washington, DC
Dhir S, Zaid AA, Hallan V (2013) Molecular characterization and recombination analysis of the complete genome of apple chlorotic leaf spot virus. J. Phytopathol. 161:704–712
Gilmer R, Mink 01, Shay JR, Stouffer RF, McCrum RC. 1971. Latent viruses of apple. I. Detection with woody indicators. Search (Agric) 1(10):1-21. NY State Agric Exp St, Geneva, NY
Komatsu K, Hirata H, Fukagawa T, Yamaji Y, Okano Y, IshikawaK Adachi T, Maejima K, Hashimoto M, Namba S (2012) Infection of capilloviruses requires subgenomic RNAs whose transcription is controlled by promoter-like sequences conserved among flexiviruses. Virus Res. 167:8–15
Marais A, Faure C, Mustafayev E, Barone M, Alioto D, Candresse T (2015) Characterization by deep sequencing of Prunus virus T, a novel tepovirus infecting Prunus species. Phytopathology 105:135–140
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
Nichols LP. 1986. Disease-resistant crab apples. Plant pathology Contrib 1558. Penn State Univ, UnivPark
Roossinck M (1997) Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 35:191–209
Simon AE, Bujarski JJ (1994) RNA-RNA recombination and evolution in virus-infected plants. Annu. Rev. Phytopath. 32:337–362
Sztuba-Solińska J, Urbanowicz A, Figlerowicz M, Bujarski JJ (2011) RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 49:415–443
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30:2725–2729
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680
Wang M, Dai H (2015) First Report of Apple chlorotic leaf spot virus in Hawthorn in China. Plant Dis. 99:164
Worobey M, Holmes EC (1999) Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 80:2535–2543
Wu QF, Ding SW, Zhang YJ, Zhu SF (2015) Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Annu. Rev. Phytopathol. 53:425–444
Yaegashi H, Isogai M, Tajima H, Sano T, Yoshikawa N (2007) Combinations of two amino acids (Ala40 and Phe75 or Ser40 andTyr75) in the coat protein of Apple chlorotic leaf spot virus are crucial for infectivity. J. Gen. Virol. 88:2611–2618
Zerbino DR, Birney E (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829
Zhu H, Wang GP, Hu HJ, Tian R, Hong N (2014) The genome sequences of three isolates of Apple chlorotic leaf spot virus from pear (Pyrus sp.) in China. Can. J. Plant Pathol. 36:396–402
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This research was financially supported by the Beijing Municipal Commission of Education (KM201610020008) and the support program of General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China (201310068, 2015IK022).
Conflict of interest
All the authors declare that we have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Li, Y., Deng, C., Bian, Y. et al. Characterization of apple stem grooving virus and apple chlorotic leaf spot virus identified in a crab apple tree. Arch Virol 162, 1093–1097 (2017). https://doi.org/10.1007/s00705-016-3183-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3183-2