
Abstract
The matrix protein 2 (M2) is a spliced product of segment 7 genome of influenza A virus. Previous studies indicate its role in uncoating of the viral ribonucleoprotein complex during viral entry and in membrane scission while budding. Despite its crucial role in the viral life cycle, little is known about its subcellular distribution and dynamics. In this study, we have shown that the M2 protein is translocated from the membrane to the cytoplasm by a retrograde route via endosomes and the Golgi network. It utilizes retromer cargo while moving from the endosome to the trans-Golgi network and prevents endosome fusion with the lysosome. Further, M2 interacts with the endoplasmic-reticulum-resident AAA-ATPase p97 for its release into the cytoplasm. Our study also revealed that the M2 protein in the cellular milieu does not undergo ubiquitin-mediated proteasomal degradation. The migration of M2 through this pathway inside the infected cell suggests possible new roles that the M2 protein may have in the host cytoplasm, apart from its previously described functions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The eukaryotic secretory system is highly organized and tightly regulated through a series of endomembrane organelles. The constant outward flux of lipids and proteins in anterograde transport vesicles is compensated by the influx of materials through retrograde transport vesicles [1]. Retrograde transport is broadly described as the inward flow of proteins and lipids from plasma membrane to Golgi and further to the endoplasmic reticulum (ER) [2]. Proteins shuttling as retrograde cargo are exemplified by acid hydrolases like vacuolar protein sorting 10 from budding yeast S. cerevisiae [3] and mammalian protein mannose 6-phosphate receptor (MPR, both cation dependent and independent) [4]. Retrograde trafficking encompasses movement to the Golgi while passing through early and/or recycling endosomes or via late endosomes [5]. Novel retrograde transport routes and cargo proteins have been identified in mammalian cells. For instance, wild-type trans-Golgi network 38 (TGN38) pass from early endosomes directly to the trans-Golgi network (TGN) [6], whereas furin pass from early endosomes to the TGN via late endosomes [7]. Interestingly, retrograde transport is exploited by various bacterial toxins and viral proteins to mediate cytotoxicity and pathogenesis, respectively. Bacterial toxins such as cholera toxin, shiga toxin (Stx) as well as the plant toxin ricin utilize this route for access to the cytoplasm [8]. Certain viral proteins rely on this pathway during infection: HIV-1 Nef protein utilizes this route for immune evasion by inducing retrograde translocation of MHC class I molecules [9]. Likewise, adeno-associated virus (AAV) 5 also exploits this pathway [10]. Each protein uses a different vesicular transport route through endosomal compartments.
Influenza A virus has a segmented, single-stranded, negative-sense RNA genome [11], and eight segments of the viral genome encode 11 proteins [12]. The M2 protein is encoded by segment 7 of influenza virus and is expressed through alternate splicing [13]. It is a type III integral transmembrane protein that forms a homotetramer. It contains three structural domains: an amino-terminal ectodomain (residues 1-24), a transmembrane domain (residues 25-43) and a cytoplasmic domain (residues 47-97) [14]. M2, although incorporated in small amounts in virions, is abundantly present in the endomembrane compartments of infected cells [15]. M2 is essential for uncoating the viral ribonucleoprotein complex inside the endosomes, and its transmembrane domain forms a pH-activated proton channel that is activated by endosomal acidification. M2 also mediates the acidification of the viral matrix and its dissociation [16]. An additional function of the M2 protein is to modify membrane curvature during virus budding. It also facilitates membrane scission, allowing the virion to be released independently of host endosomal sorting complexes required for transport (ESCRT) [17]. Another study reported that M2 aids viral pathogenesis by blocking autophagosome fusion with lysosomes, resulting in the inhibition of macroautophagy [18]. M2 also downregulates and decreases the activity of cystic fibrosis transmembrane conductance regulator (CFTR) [19]. All of these data indicate that M2 protein is a cytoplasmic-membrane-bound multi-functional protein with a variety of established cellular functions.
Here, we report that the M2 protein, like many other viral proteins and bacterial toxins, undergoes retrograde transport and can also be detected in the cytoplasm. We used an antibody internalization assay to study the trafficking of the M2 protein in host cells to avoid the effect of virus proliferation and possibly triggering the host cells’ innate immune response. In the present study, we show that the M2 protein is translocated from the membrane to the cytoplasm by a retrograde route via endosomes and the Golgi network. It utilizes retromer cargo while moving from the endosome to the trans-Golgi network and prevents fusion of endosomes with lysosomes. Further, M2 interacts with the endoplasmic-reticulum-resident AAA-ATPase p97 for its release into the cytoplasm. We also show that the M2 protein in the cellular milieu is resistant to ubiquitin-mediated proteasomal degradation. These data suggest that retrograde transport of M2 may play a role in generating resistance to M2-inhibiting anti-influenza drugs.
Materials and methods
Plasmid constructs
Cloning of the M2 gene of influenza A virus [A/Puerto Rico/8-SV14/1934(H1N1)] was done in pCDNA 3.1(-) his myc (Invitrogen) using the reverse transcription method. The ER retention mutant was prepared by modifying the C-terminal sequence of SIEL to KDEL by site-directed mutagenesis [20]. The clones were verified by sequencing. Wild-type and ATPase-dead p97 expression constructs (p97 and QQ p97) [21] were generous gifts from Dr. T. A. Rapoport (Department of Cell Biology, Harvard Medical School, Boston, MA, USA). Golgi and ER localization markers were obtained from Clontech Laboratories, USA (EYFP-Golgi and DsRed-ER plasmids, respectively). The endosomal marker and EGFP-Rab5 [22, 23] were kind gifts from Dr. S. Jameel (Department of Virology, ICGEB, New Delhi, India). SNX1-GFP and SNX4-GFP were obtained from CliniSciences, France.
Cell culture and drug treatment
A549 (adenocarcinomic human alveolar basal epithelial) and Huh-7 (human hepatocellular carcinoma) cells were used for infection and transfection experiments, respectively. All cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, HyClone) supplemented with 10% fetal calf serum (FCS, HyClone) and 100 units penicillin-streptomycin solution (Invitrogen) per ml. The effective concentrations of different inhibitors are cycloheximide at 20 µg/ml (Amresco), NH4Cl at 10 µM (Sigma), and MG132 at 10 µM (Calbiochem). LysoTracker (Molecular Probes) was used at a concentration of 50 nM.
Transfection and infection assays
Cells were transfected with Lipofectamine 2000 reagent (Invitrogen) and maintained in DMEM without serum or antibiotic. After 5 h of transfection, the medium was replaced with fresh DMEM supplemented with serum and antibiotic. The virus strain used for infection was influenza A virus [A/Puerto Rico/8-SV14/1934(H1N1)]. A fully confluent monolayer of A549 cells was infected with influenza virus at a multiplicity of infection (MOI) of 1 for 1 h in DMEM medium supplemented with 0.3% BSA (GIBCO). After 1 h of incubation, the cells were washed once with DMEM and then grown in DMEM supplemented with 0.3% BSA and 1 µg of N-p-tosyl-1-phenyl alanine chloromethyl ketone (TPCK) (Sigma Aldrich) per ml.
Western blot analysis and antibodies
Cells were lysed in RIPA buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) supplemented with 1X protease inhibitor cocktail (Roche) and subjected to SDS-PAGE. Antibodies against M2, GAPDH, β-actin, p97, ubiquitin and Myc tag were obtained from Santa Cruz Biotechnology, and antibodies against PDI was obtained from Cell Signaling Technology.
Immunoprecipitation assay
Cells were lysed in buffer (20 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100 with protease inhibitor cocktail) and incubated overnight with the primary antibodies listed above at 4 °C. The immunocomplexes were pulled down with protein A and G beads for 2 h, washed, and then subjected to SDS-PAGE and western blotting.
Immunofluorescence microscopy
After infection or transfection for 24 h or 48 h, cells growing on cover slips were fixed with 2% paraformaldehyde for 20 min at room temperature. They were permeabilized with 0.4% Triton X-100 for 20 min at room temperature and blocked with PBS containing 0.5% bovine serum antigen (BSA). Immunostaining was performed with the appropriate primary antibody. Unbound antibody was washed away with PBS, and cells were incubated with Alexa 488–tagged goat anti-rabbit or Alexa 594–tagged goat anti-mouse antibodies (Invitrogen). Nuclei were stained with DAPI, and slides were mounted using ProLong Antifade (Invitrogen). Photomicrographs were captured at 60X magnification using a Nikon A1R confocal microscope. Images were processed using NIS Elements AR 3.0 software (Nikon).
Antibody internalization assay
Huh-7 or A549 cells were washed twice with PBS after transfection and infection respectively. The surface receptors were then blocked using 1% BSA (Amersham) for 30 min at 37 °C, followed by incubation with anti-M2 (14C2) monoclonal antibody (0.25 µg/ml) for 45 min at 37 °C. Unbound antibody was removed by washing, and the cells were transferred to fresh DMEM for internalization of the antibody. The cells were then fixed at different times, treated with secondary antibody after permeabilization and blocking, and processed for confocal imaging.
In vivo ubiquitination assay
Prior to lysis, the cells were treated with 10 µM MG132 for 4 h. Harvested cells were lysed, and the lysates were immunoprecipitated with either a control IgG or anti-M2 IgG. The precipitates were then analyzed by western blot with anti-ubiquitin and anti-M2 monoclonal IgG antibodies.
Membrane fractionation
Membrane and cytoplasmic fractions was separated using a Qproteome Cell Compartment Kit (QIAGEN). The fractions were then subjected to SDS-PAGE analysis.
Results
M2 channels through the retrograde route to enter the cytoplasm
Given the multi-functionality and abundance of the M2 protein in cells infected with influenza A virus (IAV), we sought to dissect its trafficking in host cells. We devised an assay to explore the tropism of M2 in retrograde transport. To investigate the transport of the M2 protein, the Huh-7 cell line was used due to its high transfectability and discernable morphology for distinguishing various subcellular organelles. For monitoring the movement of M2, an antibody internalization assay was developed in which an antibody specific for the N-terminal epitope of M2 was ectopically added to live cell cultures and allowed to internalize at different time points. The assay was tested to confirm the movement of M2 by its binding to the ectopically added antibody inside the cells. After confirming its entry into the cell, the kinetics of the subsequent intracellular movement of M2 protein were investigated using fluorescent tagged plasmids for distinguishing cell organelles. First, the kinetics of localization of M2 with Rab5-GFP were examined (Fig. 1). Rab5 GTPase marks the boundary of early endosomes, phagosomes and caveosomes, hence imparting a scattered appearance inside the cell [24]. After 2.5 min, the M2 protein (red) was detected bound to the cell surface before being internalized. In some cells, a small amount of the M2 protein was observed in the peripheral puncta, which corresponded to early endosomes colocalizing with Rab5-GFP (green) (Fig. 1a, top row). At 20 min post-internalization, M2 was found in the juxtanuclear position in the cells colocalizing with Rab5-GFP (Fig. 1a, second row). The co-localization increased to maximum magnitude at 40 min, revealing that it was primarily located inside of endocytic compartments (Fig. 1a, third row). The colocalization of M2 with Rab5-GFP decreased at 1 h post-internalization (Fig. 1a, fourth row) and was observed to shift out of the endocytic vesicles to move to other compartments.

Tracing the internalization of M2 through the retrograde pathway. Huh-7 cells transiently expressing M2 with different organelle marker proteins were incubated with anti-M2 monoclonal antibody at 37 °C and then fixed at different time points. For monitoring retrograde transport of the M2 protein, an antibody internalization assay was performed. The scale bar in each figure represents 20 µm. a) Colocalization of internalized M2 protein with endosome marker Rab5-GFP. M2 antibody (red, middle panels) was labelled with Alexa 594–conjugated secondary antibody along with Rab5-GFP (green, left panels). M2 protein colocalization with Rab5-GFP is shown 1 h post-internalization (merged panels). b) Colocalization of M2 with Golgi Marker Golgi-YFP. Colocalization of Golgi-YFP (green, left panels) and M2 (red, middle panels) is shown at different time points. c) Colocalization of M2 with ER marker dsRedER. M2 antibody labelled with Alexa–488 conjugated secondary antibody (green, middle panels) and dsRedER-ER marker (red, left panels) shows M2 colocalization in the ER. d) Movement of M2 to the cytosol at different time points after internalization. The cytoplasm is labelled with anti-actin antibody and Alexa–488 conjugated secondary antibody (green, left panels)
Next, we tracked the movement of M2 to Golgi vesicles. At 3 minutes, consistent with our observation with the Rab5-GFP marker, M2 (red) was present in the peripheral puncta, corresponding to early endosomes (Fig. 1b, top row). At this point, M2 did not appear to be located in the Golgi complex (green), indicating that M2 did not directly enter the trans-Golgi network (TGN). At 1 h after entering the cells, the M2 protein partially colocalized with the Golgi marker [25] (Fig. 1b, second row), which demonstrated the beginning of its flow to the Golgi complex. At 2 h post-entry, the amount of M2 present in the Golgi reached a maximum (Fig. 1b, third row), while M2 localization decreased at 4 h post-internalization, indicating its transfer out of the Golgi (Fig. 1b, fourth row).
Subsequently, we examined the entry of M2 into the ER compartment. The M2 protein (green) did not enter into the ER (red) at 1 h post-internalization (Fig. 1c, top row) and was present in trace amounts in the ER compartment at 2 h post-internalization. Its levels peaked within the ER at 4 h post-internalization (Fig. 1c, second and third rows) and at 6 h post-internalization, the amount of M2 in the ER began to decrease and it appeared to disperse in the entire cell (Fig. 1c, fourth row).
We next studied the kinetics of M2 with actin (cytoplasmic marker) and found that the M2 protein (red) was maximally localized with actin (green) at 6 h after internalization and thus appeared to be dispersed throughout the entire cytoplasm (Fig. 1d, second row). At the 4 h and 8 h time points, M2 protein (red) was localized near or around the nucleus and had not spread to the entire cell (Fig. 1d, first and third rows). Through these systematic studies we were thus able to track the M2 protein from the cell surface to the endosomes, Golgi and ER over time.
To investigate a similar scenario in the case of influenza virus infection, A549, cells after infection with the IAV (H1N1/PR8) strain, were subjected to a similar assay for monitoring M2 at different time points after infection. Upon infection, M2 was first detected to be internalized inside the cell at 4 h postinfection (Fig. S1). This was not observed in the first 1-2 h postinfection, which could be attributed to a lag in protein synthesis. However, when followed up to 24 h, M2 was found to be internalized into the cell at each time point after 5 h postinfection (not shown).
M2 progresses as retromer cargo while moving from endosomes to the trans-Golgi network, but it does not reach the lysosomes
Proteins from endosomes move to the trans-Golgi network (TGN) through retromer cargo. This cargo is marked by signature protein sub-complexes such as sorting nexins (SNX), which forms transport carriers and guides the protein to TGN [26]. There are 12 SNX proteins named SNX 1 to 12 [27]. We analyzed the association of M2 with a few SNX proteins over time. Huh7 cells were co-transfected with GFP-tagged SNX-1 and SNX-4 and M2 expressing plasmids and were subjected to the antibody internalization assay. Both SNX1-GFP and SNX4-GFP were colocalized with M2 protein when traced from early stages of transfection until 4 hr post-internalization (Fig. 2a and b). This explicitly demonstrated the movement of M2 through the retrograde transport pathway.

M2 progress from endosome to trans-Golgi as retromer cargo. a) M2 colocalization with SNX1 while it is moving from endosome to Golgi. Huh7 cells cotransfected with SNX1-GFP and M2 were processed for antibody internalization assay as described in Materials and methods. b) M2 colocalization with SNX4 and its movement from endosome to Golgi. Huh7 cells cotransfected with SNX4-GFP and M2 were processed for antibody internalization assay. Colocalization of SNX4 (green, left panels) and M2 (red, middle panels) is seen at different time points after internalization. c) M2 follows the classical retrograde pathway and does not deviate to the lysosome. M2-transfected cells were treated with LysoTracker and subjected to antibody internalization assay
The vesicular transport pathway has a variety of exit routes from endosomes [2]. One of the exit routes is passage to lysosomes, where proteins are degraded by lysosomal enzymes [18]. In order to demonstrate whether the M2 protein moves to the lysosomes for degradation during retrograde movement, its localization with LysoTracker was investigated. Cells were transfected with the M2 expressing plasmid and subsequently subjected to antibody internalization assay. LysoTracker, which binds to lysosomes was then added to the cells. We did not see any evidence that the M2 protein enters the lysosome at least up to 4 h post-internalization (Fig. 2c). This illustrates that the M2 protein strictly follows the retrograde route and does not deviate to other vesicles.
AAA-ATPase p97 guides the movement of M2 from the ER to the cytoplasm
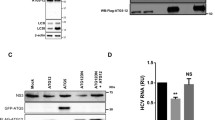
Misfolded and unfolded proteins exit to the cytosol for proteasomal degradation through the action of ATPase. The AAA-ATPase p97/VCP (ATPase associated with various cellular activities) is essential for the retrograde transport of misfolded proteins and binds to poly-ubiquitinated proteins, guiding them to proteolysis [28]. Many viral proteins, such as the ORF2 protein of hepatitis E virus, interact with p97 to gain access to the cytoplasm [22]. The M2 protein was examined for its interaction with p97 as a possible route to enter the cytoplasm. Huh7 cells co-expressing M2 with p97 or p97 QQ (ATPase mutant form) were lysed and immunoprecipitated with an anti-M2 antibody and immunoblotted with an anti-p97 IgG. The M2 protein was co-immunoprecipitated with p97 wild-type but not with the mutant ATPase (Fig. 3a). This suggests that M2 interacts with the p97/VCP protein and that the ATPase domain is necessary for maintaining this interaction.

M2 interaction with AAA-ATPase p97 protein in the ER for its retrograde transport. a) Huh7 cells co-expressing M2 with either wild-type p97 (p97 WT) or mutant p97 (p97 QQ) were lysed along with control p97 cell lysate (lane 2), followed by immunoprecipitation with anti-M2 antibody. Immunoblotting was done with anti-p97 antibody (upper panel) as well as anti-M2 antibody (lower panel). b) An ER-retained mutant of M2 (ER-M2) was coexpressed with wild-type p97 or mutant p97, and cell lysates were made and compared with a mock-transfected cell lysate (lane 1). The samples were immunoprecipitated with anti-M2 antibody and immunoblotted with anti-p97 antibody (upper panel) and anti-M2 antibody (lower panel). * indicates the light-chain band. c) Infected A549cells were harvested after 24 h and immunoprecipitated with either anti-p97 antibody (upper panels) or anti-M2 antibody (lower panels). The immunoprecipitates were immunoblotted with both anti-p97 and anti-M2 antibody. d) WT and mutant (QQ) forms of p97 were co-expressed with M2, and the movement of M2 was traced at 6 h post internalization. M2 antibody was labelled with Alexa 594 secondary antibody, and actin (cytoplasmic marker) was labelled with Alexa 488 secondary antibody. e) WT p97 was co-expressed with M2, and the movement of M2 in the ER and cytoplasm was traced at 6 h post-internalization. Co-localization in the ER (upper panel) and colocalization with actin, a cytoplasmic marker (lower panel), are shown. DsRedER was used as an ER marker and M2 antibody was labelled with Alexa 488 secondary antibody. f) A mutant of p97 was co-expressed with M2, and the movement of M2 in the ER and cytoplasm was traced at 6 h post-internalization
Since p97 interacted with M2, it was postulated that this may play a role in the movement of M2 from the ER to the cytosol. The wild-type and mutant p97 were co-expressed along with M2, and the localization of retrograde-transported M2 protein was examined at 6 h post-internalization. M2 with the wild type-p97 (WT-p97) exhibited a dispersed distribution when compared with the mutant-p97 (QQ-p97), which displayed restricted localization around the nucleus (Fig. 3b). The localization of retrograde-transported M2 in the ER was resolved at 6 h post-internalization.
The interaction of M2 with p97 was also confirmed in influenza-virus-infected cells. The infected A549 cells were lysed and immunoprecipitated with both anti-M2 and anti-p97 antibodies (Fig. 3c). The cells were immunoblotted with anti-p97 and anti-M2 antibody to detect the co-immunoprecipitation. The p97 protein was co-immunoprecipitated with M2 antibody (Fig. 3c). This indicated that M2 and p97 interacted in the infection system as well. These results indicated that M2 interacts with the p97 protein of the ER and utilizes it as a channel for entry into the cytoplasm.
We observed that the co-expression of wild-type p97 with M2 protein maximizes its expression but results in a dispersed distribution and co-localization with both actin (Fig. 3d), a cytoplasmic marker, and an ER marker (Fig. 3e). With the QQ mutant of p97, M2 was limited to the ER (Fig. 3f) as its degree of co-localization with the ER marker was greater than that of its co-localization with actin (Fig. 3e and f). This suggests that the ATPase mutant is unable to transfer M2 from the ER to the cytosol, that the wild-type ATPase p97 molecule is required for the exit of M2 from the ER to the cytoplasm, and that M2 moves into the cytoplasm in an AAA-ATPase-p97-dependent manner.
Fate of M2 in cytoplasm: stability analysis
Retro-translocated proteins migrating through the endoplasmic reticulum-associated-protein degradation (ERAD) pathway are ideal substrates for rapid 26S-proteasome-mediated degradation [29]. In order to determine the half-life of M2 protein in the cells, a cycloheximide chase assay was performed in Huh7 cells. This experiment revealed that even after 15 h of cycloheximide (CHX) treatment, a small fraction of the M2 protein was still observed in the cells (Fig. 4a). Band intensity was analyzed to determine its half-life, normalized to GAPDH, which was found to be 12 h. Next, to check if the small fraction of M2 is retrograde transported from the cell surface to the cytoplasm, Huh7 cells expressing control or M2 protein were treated with 20 µg of CHX per ml and the membrane/cytoplasmic fractions were separated (Fig. S2). We also examined whether the M2 protein gets ubiquitinylated after moving into the cytoplasm. This was done by treating cells with MG132 (a proteasome inhibitor) [30] prior to cell lysis in order to increase the quantity of ubiquitinylated proteins. More ubiquitinylated bands were observed with the control IgG when compared to those immunoprecipitated with the M2 antibody (Fig. 4b). Since there was no M2 equivalent band, these bands could be unrelated proteins pulled down nonspecifically by the antibody. In summary, M2 protein does not appear to be ubiquitinylated. Finally, we tested the effects of lysosomal (NH4Cl) and proteasomal (MG132) inhibitors on the stability of M2 as described previously [22]. After 16 h of cycloheximide chase, MG132 had negligible effect on the stability of M2, while NH4Cl stabilized M2 to approximately 40 percent (Fig. 4c). This indicated that M2 is not a substrate of the proteasome complex and confirmed that ubiquitin-proteasome degradation pathways are not responsible for degradation of the viral M2 protein.

Stability and degradation of M2 inside the cytoplasm. a) Half-life analysis of the M2 protein expressed in Huh7 cells was done using cycloheximide chase at a concentration of 20 µg/ml. The graph represents densitometric analysis showing the half-life of M2 protein normalized to GAPDH. b) Determination of ubiquitination of M2 by immunoprecipitation (IP) with anti-M2 antibody or IgG and immunoblotting (IB) with anti-ubiquitin (upper panel) as well as anti-M2 antibody (lower panel). All samples were treated with MG132 (10 µM) 3 h prior to cell lysis. Ten percent of the lysate was kept for input control. * indicates the light-chain band. c) At 16 h after cycloheximide (CHX) treatment, M2-expressing cells were treated with MG132 or NH4Cl (10 µM) to investigate the mode of degradation of M2 3 h prior to lysis. Bands were quantified using ImageJ software as shown in the lower panel. Error bars are SEM of analyses of 3-4 blots
Discussion
Influenza virus has been the cause of many recurrent epidemics and pandemics for centuries [31]. Even with progress in modern medicine and with advent of various new drugs and vaccines, influenza virus has an ability to mutate and evolve to become resistant to previously effective therapy. Hence, there is a constant need to develop new drugs to prevent and treat influenza infections. Although many aspects of influenza virus pathogenesis have been elucidated, an extensive understanding of host-pathogen interactions and many aspects of viral proteins that utilize host cells and may contribute to pathogenesis of the virus remain to be identified. New understanding of viral pathogenesis is required for effective strategies to develop novel pharmaceutical therapies. Influenza virus encodes 11 proteins, all of which appear to have multiple functions [11, 12]. M2 is an ion channel protein and is considered to play crucial roles in the viral life cycle. M2 has been shown to play a role in autophagy, in restricting autophagosome fusion with lysosomes, and in causing membrane scission during virus budding through an ESCRT-independent pathway [16–18]. Here, we show a novel property and tropism of the M2 protein associated with its progression through retrograde transport and elucidate the kinetics of the retrograde motion of M2 utilizing an already known vesicular transport pathway (Fig. 1).
The M2 protein is abundantly present in influenza-virus-infected cells, while approximately 15 particles are incorporated into the newly budded virion. This pool of proteins is presumed to play a significant role in the influenza virus life cycle. M2 has been reported previously to cause membrane scission during influenza virus assembly by bringing about membrane ruffling. In our study, it was shown that M2, like other bacterial toxins or viral proteins, tends to undergo retrograde transport and hence flow in the backward motion from the membrane to the cytosol through various organelles. When the kinetics of M2 localization were studied with each organelle marker, an orderly temporal flow through the vesicular transport pathway was observed (Fig. 1). The exit of M2 from the ER to the cytoplasm is a short-lived process and occurs within a short time frame, after which M2 either flows back to ER or is degraded (Fig. 1D). Infection with influenza virus demonstrated that the dynamics of the flow of M2 corresponded to what was observed using the transfection system (Fig. S1). This confirms that our transfection system mimics the events occurring during live virus infection, and infection with live influenza virus also resulted in the same retrograde phenomenon, even under the influence of other viral proteins. However, since infecting cells with influenza A virus initiates a complex virus replication process and possibly triggers innate immune responses by the host cells, our transfection system made it possible to study the tropism of M2. The retrograde transport of M2 represents a pathway that is important in the influenza virus life cycle that might be utilized for designing novel anti-influenza medicines.
Pathogens exploit more than one endocytotic pathways for faster migration through the cell and for promotion of pathogenesis [32]. One of the important protein complexes distinguishing retrograde transport from other vesicular trafficking is the retromer complex [33]. This is a group of proteins that bind to and guide the cargo destined for movement from early endosomes to the TGN. Retromers consist of two subcomplexes, the vacuolar protein sorting (vps) heterotrimer and the sorting nexin [27]. In this study, we found that SNX proteins are involved in guiding the M2-bearing retrograde cargo (Fig. 2). Furthermore, the demonstration of an interaction of M2 with wild-type p97 ATPase corroborates our hypothesis that it migrates to the cytoplasm through retro-translocation, as M2 recruits p97 to transport it from the ER to the cytoplasm (Fig. 3). To our knowledge, this is the first report on the transport of the M2 protein through the cytoplasm and its interaction with actin.
Many viral proteins have few lysine residues in order to avoid ubiquitination and subsequent degradation. The amino acid sequence, when analyzed using software for identifying probable ubiquitinylation sites revealed the absence of such moieties [29]. This was confirmed using an in vivo ubiquitination assay that showed the absence of poly-ubiquitin molecules attached to M2. Our study reveals the new phenomenon of retrograde transport adopted by influenza virus M2 protein, which opens a new avenue to understanding viral pathogenesis and will pave the way for effective interventions and strategies to combat influenza infections.
References
Lodish H, Berk A, Matsudaira P et al (2003) Molecular Cell Biology, 5th edn. W.H.Freeman, New York, pp 701–742
Bonifacino JS, Rojas R (2006) Retrograde transport from endosomes to the trans- Golgi network. Nat Rev Mol Cell Biol 7:568–579
Pfeffer SR (2001) Membrane transport: retromer to the rescue. Curr Biol 11:R109–R111
Ghosh P, Dahms NM, Kornfeld S (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4:202–213
Johannes L, Popoff V (2008) Tracing the retrograde route in protein trafficking. Cell 135:1175–1187
Kuwana T, Peterson PA, Karlsson L (1998) Exit of major histocompatibility complex class II—invariant chain p35 complexes from the endoplasmic reticulum is modulated by phosphorylation. Proc Natal Acad Sci 95:1056–1061
Bosshart H, Humphrey J, Deignan E, Davidson J, Drazba J et al (1994) The cytoplasmic domain mediates localization of furin to the trans-Golgi network en route to the endosomal/lysosomal system. J Cell Biol 126:1157–1172
Falnes PO, Sandvig K (2000) Penetration of protein toxins into cells. Curr Opin Cell Biol 12:407–413
Johannes L, Pezo V, Mallard F, Tenza D, Wiltz A et al (2003) Effects of HIV-1 Nef on retrograde transport from the plasma membrane to the endoplasmic reticulum. Traffic 4:323–332
Dales S (1975) Part VII. Microtubules and cell surface organization: involvement of the microtubule in replication cycle of animal viruses. Ann NY Acad Sci 253:440–444
Cheung TK, Poon LL (2007) Biology of influenza A virus. Ann NY Acad Sci 1102:1–25
Lamb R (1983) The influenza virus RNA segments and their encoded proteins. Genetics of influenza viruses. Springer, New York, pp 21–69
Fischer WB, Hsu HJ (2011) Viral channel forming proteins: modeling the target. BBA Biomembr 1808:561–571
Pinto LH, Lamb RA (2006) The M2 proton channels of influenza A & B viruses. J Biol Chem 281:8997–9000
Henkel JR, Apodaca G, Altschuler Y, Hardy S, Weisz OA (1998) Selective perturbation of apical membrane traffic by expression of influenza M2, an acid-activated ion channel, in polarized Madin–Darby canine kidney cells. Mol Biol Cell 9:2477–2490
Takeda M, Pekosz A, Shuck K, Pinto LH, Lamb RA (2002) Influenza a virus M2 ion channel activity is essential for efficient replication in tissue culture. J Virol 76:1391–1399
Rossman JS, Jing X, Leser GP, Lamb RA (2010) Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913
Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T et al (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380
Londino JD, Lazrak A, Jurkuvenaite A, Collawn JF, Noah JW et al (2013) Influenza matrix protein 2 alters CFTR expression and function through its ion channel activity. AM J Physiol Lung C 304:L582–L592
Munro S, Pelham HR (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell 48:899–907
Ye Y, Meyer HH, Rapoport TA (2001) The AAA-ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414:652–656
Surjit M, Jameel S, Lal SK (2007) Cytoplasmic localization of the ORF2 protein of hepatitis E virus is dependent on its ability to undergo retrotranslocation from the endoplasmic reticulum. J Virol 81:3339–3345
Surjit M, Varshney B, Lal SK (2012) The ORF2 glycoprotein of hepatitis E virus inhibits cellular NF-kappa B activity by blocking ubiquitination mediated proteasomal degradation of I-kappa B alpha in human hepatoma cells. BMC Biochem 13:7–14
Stenmark H, Olkkonen VM (2001) The Rab GTPase family. Genome Biol 2:S3007
Saraste J, Palade GE, Farquhar MG (1987) Antibodies to rat pancreas Golgi subfractions: identification of a 58-kD cis-Golgi protein. J Cell Biol 105:2021–2029
Rojas R, Kametaka S, Haft CR, Bonifacino JS (2007) Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol Cell Biol 27:1112–1124
Wassmer T, Attar N, Bujny MV, Oakley J, Traer CJ et al (2007) A loss-of-function screen reveals SNX5 and SNX6 as potential components of the mammalian retromer. J Cell Sci 120:45–54
Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19:2181–2192
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose regulated proteins. Nature 332:462–464
Han YH, Moon HJ, You BR, Park WH (2009) The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep 22:215–221
Taubenberger JK, Morens DM (2010) Influenza: the once and future pandemic. Public Health Rep 3:16–26
Marsh M, Helenius A (2006) Virus entry: open sesame. Cell 124:729–740
Bujny MV, Popoff V, Johannes L, Cullen PJ (2007) The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. J Cell Sci 120:2010–2021
Acknowledgements
We thank Dr. T. A. Rapoport and Dr. S. Jameel for providing us the plasmid constructs. Our special thanks also go to R. Kumar and P. Kumar for assistance in virus infection, confocal microscopy and cell culture experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Bhowmick: Deceased.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bhowmick, S., Chakravarty, C., Sellathamby, S. et al. The influenza A virus matrix protein 2 undergoes retrograde transport from the endoplasmic reticulum into the cytoplasm and bypasses cytoplasmic proteasomal degradation. Arch Virol 162, 919–929 (2017). https://doi.org/10.1007/s00705-016-3153-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3153-8