
Abstract
Bovine leukemia virus (BLV) is the etiological agent of enzootic bovine leukosis, which is the most common neoplastic disease of cattle. BLV infects cattle worldwide and affects both health status and productivity. However, no studies have examined the distribution of BLV in Myanmar, and the genetic characteristics of Myanmar BLV strains are unknown. Therefore, the aim of this study was to detect BLV infection in Myanmar and examine genetic variability. Blood samples were obtained from 66 cattle from different farms in four townships of the Nay Pyi Taw Union Territory of central Myanmar. BLV provirus was detected by nested PCR and real-time PCR targeting BLV long terminal repeats. Results were confirmed by nested PCR targeting the BLV env-gp51 gene and real-time PCR targeting the BLV tax gene. Out of 66 samples, six (9.1 %) were positive for BLV provirus. A phylogenetic tree, constructed using five distinct partial and complete env-gp51 sequences from BLV strains isolated from three different townships, indicated that Myanmar strains were genotype-10. A phylogenetic tree constructed from whole genome sequences obtained by sequencing cloned, overlapping PCR products from two Myanmar strains confirmed the existence of genotype-10 in Myanmar. Comparative analysis of complete genome sequences identified genotype-10-specific amino acid substitutions in both structural and non-structural genes, thereby distinguishing genotype-10 strains from other known genotypes. This study provides information regarding BLV infection levels in Myanmar and confirms that genotype-10 is circulating in Myanmar.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Species Bovine leukemia virus belongs to family Retroviridae, is an oncogenic member of genus Deltaretrovirus, and its isolates are a model of pathogenesis for human T cell leukemia virus type 1 (HTLV-1) [1]. Bovine leukemia virus (BLV) is the causative agent of enzootic bovine leukosis (EBL), the most common neoplastic disease of cattle [2, 3]. Symptoms/signs of lymphoma caused by BLV infection depend on the site of the tumors, but include digestive disturbance, loss of appetite, weight loss, weakness, reduction of milk production or general debility, and various neurological manifestations [4]. The complete BLV genome comprises structural and enzymatic gag, pro, pol, and env genes, regulatory genes tax and rex, and accessory genes R3 and G4 [2, 3]. The BLV gag gene encodes structural proteins that play a key role in viral assembly and genome packaging [5]. The BLV pro and pol genes encode proteases (PRT) p14, and p80 (RT/IN) which has reverse transcriptase (RT), RNAse H and integrase activity (IN), respectively [3]. The regulatory gene tax encodes the Tax protein, which plays an essential role in transformation during BLV-induced leukemogenesis [6] and regulates viral expression at the transcriptional level [3]. Another regulatory gene, rex, encodes the Rex protein, which is responsible for nuclear export of viral RNA and accelerates cytoplasmic accumulation and translation of viral messenger RNA (mRNA) in BLV-infected cells [7]. The R3 and G4 proteins help to maintain a high viral load [8, 9].
Like other structural genes, the BLV env gene is necessary for the synthesis of virions. The env gene is transcribed as a 5.1 kb mRNA encoding the pr72env precursor [10, 11], which is then cleaved to yield the extracellular gp51 and transmembrane gp30 proteins. Among the Env glycoproteins, gp51 plays an essential role in the viral life cycle [12–14] and is required for cell entry; it is also the target of neutralizing antibodies [12, 14, 15]. The N-terminal half of BLV gp51 contains three conformational epitopes, F, G, and H [16], and plays an important role in viral infectivity and syncytium formation [15, 16], while the C-terminal half contains the linear epitopes A, B, D, and E [10, 16]. Therefore, the gp51 region is widely used for BLV genotyping studies.
Previous phylogenetic studies of this region from viral strains isolated worldwide demonstrate that BLV can be classified into at least eight genotypes [17–28]. Furthermore, a recent study that focused on the genetic variability of BLV strains in South America by analyzing full genome sequences revealed the existence of a novel genotype (genotype-9) in Bolivia, indicating that BLV is more diverse than previously thought [29]. A very recent study of the molecular epidemiology and serology of BLV infection in Thailand identified another novel BLV genotype, named genotype-10, indicating that at least ten different genotypes of BLV are circulating worldwide [30].
Myanmar is the largest country in mainland Southeast Asia. Its economy is based on agriculture. Among all livestock in Myanmar, cattle comprise the main population, although there are no beef cattle. Since Myanmar is an agro-based country, draught cattle (beasts of burden) are accorded the highest priority in the livestock program. Dairy cattle play a secondary role in ruminant production, although their use has increased due to demand for milk and milk products by urban populations and improved nutritional standards and quality of the life of the rural population [31]. A variety of BLV genotypes have been detected in different parts of Asia: genotype-1, -2, and -3 in Japan [23, 32], genotype-1 and -6 in the Philippines [26], genotype-1, and -3 in Korea [27], genotype-1, -4, and -7 in Mongolia [28], and genotype-1, -6, and -10 in Thailand [30]. However, the prevalence of BLV infection and the existence of EBL in this region are unknown [4].
Here, we examined the distribution and molecular characteristics of BLV strains in Myanmar and compared them with those of other BLV strains worldwide. First, we tested blood samples obtained from 66 cattle in Myanmar using two methods that target the BLV long terminal repeat (LTR) region, namely, nested polymerase chain reaction (PCR) and real-time PCR (specifically, BLV-CoCoMo-qPCR-2). The results were confirmed using nested PCR targeting the BLV env-gp51 region and real-time PCR targeting the BLV tax gene. Second, the partial and full-length env-gp51 sequences of five Myanmar BLV strains were used for phylogenetic analysis and compared with isolates from other geographical locations worldwide. Third, the full genome sequences of two Myanmar BLV strains were obtained and their genetic variability and genotype were analyzed together with those of 27 BLV whole genome sequences from the NCBI database. In summary, this study aimed to examine the prevalence and molecular characteristics of BLV genotypes in Myanmar.
Materials and methods
Sample collection and extraction of genomic DNA
Blood samples were obtained from 66 cattle on 23 farms in four townships of the Nay Pyi Taw Union Territory in central Myanmar, namely, Zeyar thiri Township, Tatkone Township, Pyinmana Township, and Lewe Township (Table 1). These cattle are of Pyar Sein breed, Friesian, Jersey and Pyar Ni breeds, with ages ranging from the youngest cattle, three-months old, to the oldest, 11-years old. All of the farms are small-holding farms, having less than 10 cattle in each farm. Genomic DNA was extracted from the buffy coat separated from whole blood using the standard phenol-chloroform method [33].
Detection of BLV provirus by nested PCR and real time-PCR targeting BLV LTR regions
BLV LTR regions were detected by nested PCR [26, 29] and BLV-CoCoMo-qPCR-2 [26, 34–36], as described previously.
Detection of BLV provirus by real-time PCR targeting the BLV tax gene
The BLV tax gene was detected using the Cycleave PCR BLV detection kit (Takara Bio Inc., Otsu, Japan), according to the manufacturer’s instructions. The kit amplifies the BLV tax gene and detects it with a FAM-labeled Cycleave probe.
Detection of BLV provirus by nested PCR targeting the BLV env-gp51 region
The BLV env-gp51 region was detected by nested PCR, as described previously [26, 29].
PCR amplification and sequencing of the partial BLV env-gp51 gene
Only samples that were positive by env-gp51 nested PCR method (L1, L2, S3, S5 and p60, but not S1) were used for further analysis in terms of successful amplification of BLV env-gp51. The partial BLV env-gp51 gene was amplified from five BLV-positive samples by nested PCR using PrimeSTAR GXL DNA Polymerase (Takara Bio Inc.) as described for the nested PCR targeting the BLV env-gp51 region [26, 29]. Positive second-round PCR products were purified using Exo-SAP IT (USB Corp., Cleveland, OH) and sequenced on an ABI3730xl DNA Analyzer using an ABI PRISM Big Dye Terminator v 3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). Sequences included a 475 bp sequence (without primers) of the env-gp51 gene, corresponding to nucleotides 5090–5564 of the whole genome sequence of the FLK-BLV subclone pBLV913 (GenBank accession number EF600696) [37]. Editing, alignment, and identification of the nucleotide sequences were performed using MEGA 5.1 software [38].
PCR amplification, cloning of the PCR products, and sequencing of the complete env-gp51 gene
Complete BLV env-gp51 genes were amplified using KOD FX Neo (TOYOBO Co., Ltd., Osaka, Japan) and the following primer pairs: forward, pBLV-F0 5′-AGATGGGAGCTACACCATTCA-3′ and reverse, pBLV-0R 5′-GTCTGTAGAGACTCTTTGCGAG-3′. The reaction mixture contained 12.5 μl of 2× PCR Buffer for KOD FX Neo, 5 μl of 2 mM dNTP mix, 0.5 μl of KOD FX Neo Polymerase (TOYOBO Co., Ltd.), 1 μl of each primer (10 μM), and 2 μl of distilled water (to yield a final reaction volume of 25 μl). The PCR amplification conditions were as follows: 94 °C for 2 min, followed by 30 cycles of denaturation at 98 °C for 10 sec, annealing at 60 °C for 30 sec, and extension at 68 °C for 45 sec. Successful amplification resulted in a 1465 bp DNA fragment. PCR amplicons were purified using Centri-Sep Columns (Princeton Separations, Inc. Adelphia, NJ).
Because of the blunt-ended DNA termini produced by KOD FX New, the purified PCR products derived from the complete env gene were treated at 70 °C for 15 min with rTaq polymerase (TOYOBO Co., Ltd.) to add a single 3′-A overhang at both ends and then purified again using Centri-Sep Columns (Princeton Separations, Inc.), according to the manufacturer’s instructions. The rTaq treated and purified PCR products were then ligated into the pGEM-T easy vector using T4 ligase (Promega Corporation, Madison, WI), according to the manufacturer’s instructions. An aliquot of the ligation mixture (10 μl) was then transformed into E. coli XL10-Gold Ultracompetent cells (Agilent Technologies Co., Ltd. Adelphia, NJ) and incubated at 37 °C for 90 min. Next, the transformed E. coli were spread on agar plates containing 100 mg/ml ampicillin, 100 mM IPTG, and 20 mg/ml X-Gal. At least 20 white colonies were selected to obtain positive clones. The FastGene plasmid mini kit (Nippon Genetics Co., Ltd., Tokyo, Japan) was used for plasmid DNA extraction.
Three positive plasmids were confirmed by sequencing on an ABI3730xl DNA Analyzer using an ABI PRISM Big Dye Terminator v 3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems) and the following sequencing primers: pBLV-F0 plus pBLV-0R and gp51-R-5639 (5′-AWCAACAACCTCTGGGAAGGGT-3′) and pBLV-F1 (5′-TCAGAGACTCACCTCCCTG-3′). Sequences of 1230 bp (including the leader peptide, full-length gp51, and the N-terminal region of gp30) corresponding to nucleotide positions 4826–6054 of the whole genome sequence of the FLK-BLV subclone pBLV913 (GenBank accession number EF600696) [37] were obtained and deposited in the DNA Data Bank of Japan (DDBJ) database under accession numbers LC154064–LC154067.
PCR amplification, cloning of PCR products and sequencing of the complete BLV provirus genome
Because strains showed homology to each other, based on full-length env-gp51 analysis, we chose two distinctive BLV strains for subsequent sequencing. Complete BLV genome sequences from two Myanmar strains were obtained by PCR amplification of overlapping genomic fragments using PrimeSTAR GXL DNA Polymerase (Takara Bio Inc.) and specific primers (Life Technologies Japan Ltd, Tokyo, Japan) (Additional file 1: Figure S1). The final reaction mixture (25 μl) contained 5 μl of 5× PrimerSTAR GXL Buffer, 2 μl of 2.5 mM dNTP mix, 1 μl of each primer (each at 10 pmol), 2 μl of template (30 ng/μl), and 0.5 μl of PrimerSTAR GXL DNA Polymerase. PCR amplification was performed as follows: 98 °C for 2 min, followed by 33 cycles of denaturation at 98 °C for 15 sec, annealing at 60 °C for 15 sec, and extension at 68 °C for 1 min/kb (1 min per kilobase). The PCR products were purified using a FastGene Gel/PCR extraction kit (Nippon Genetics Co., Ltd., Tokyo, Japan), according to the manufacturer’s instructions.
The PCR amplicons were cloned as described for the complete BLV env-gp51 gene. Three positive plasmids were confirmed by sequencing on an ABI3730xl DNA Analyzer using an ABI PRISM Big Dye Terminator v 3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems).
Construction of the phylogenetic tree
The partial and complete BLV env-gp51 sequences from the Myanmar samples were aligned with 102 partial or 98 complete BLV env-gp51 sequences from GenBank (representative of the ten known BLV genotypes) using MEGA 5.1 software. Phylogenetic analyses of a partial (475 bp) and a complete (807 bp) sequence of the env-gp51 gene were conducted using MEGA 5.1. For robust and accurate phylogenetic analysis of the BLV env-gp51 sequences, the “find best DNA/Protein models” tool of MEGA 5.1 software was used to choose the best fit model. The Kimura-2 parameter model with gamma distribution (K2+G) was chosen as the model with the best fit to analyze the BLV env-gp51 sequences with the smallest Akaike Information Criterion value (AICc). Phylogenetic trees were constructed using the maximum likelihood (ML) algorithm with the K2+G model of nucleotide substitution [39] in Mega 5.1. The reliability of the phylogenetic relationships was evaluated by nonparametric bootstrap analysis with 1000 replicates. The complete genome sequences of Myanmar BLV strains were aligned with 27 complete BLV sequences, including previously known sequences in the NCBI database and previously identified South American strains [29], using MAFFT v.7.0 [40]. The ML tree based on BLV complete genome sequences was constructed by Mega 5.1. Deduction of protein sequences through translation of nucleotide to amino acid sequences was performed using MEGA.
Results
The spread of BLV infection in Myanmar
To investigate the spread of BLV infection in Myanmar, we collected 66 blood samples from cattle in different farms in four regions of Nay Pyi Taw Union Territory of central Myanmar. Genomic DNA isolated from these samples was first screened for BLV infection by nested PCR and BLV-CoCoMo-qPCR-2 targeting the BLV LTRs (two copies of which are present in each individual BLV genome) (Table 1). BLV-CoCoMo-qPCR-2 identified 6 out of 66 samples as positive for BLV provirus, whereas nested PCR identified five. To confirm whether the six positive animals were BLV-infected, all six samples were analyzed using PCR methods targeting the BLV tax and env-gp51 genes (Table 1). Real-time PCR targeting the tax gene identified three of the six as positive for BLV provirus, whereas nested PCR for the env-gp51 gene identified five. Only two of the six samples were positive according to all four methods. Thus, we determined that samples that were positive in at least two assays were “BLV positive”, establishing 6 out of the 66 animals as being infected.
Samples from Zeyathiri Township were all negative. Three cattle samples out of four from Tatkone Township were positive. Of the 20 samples collected from different farms in Pyinmana Township, only one sample (5.0 %) was positive. Two out of thirty-five (5.7 %) samples collected from Lewe Township were positive.
Phylogenetic analysis based on partial and complete sequences of env-gp51 of Myanmar strains
Since the partial env-gp51 region is commonly used for BLV genotyping studies [17–27], we used partial and complete env-gp51 sequences from five of the six BLV-positive samples for phylogenetic analysis. After direct sequencing, we aligned the partial (475 bp) env-gp51 nucleotide sequences of these five field strains (corresponding to nucleotide positions 5090–5564 of the whole genome sequence of the FLK-BLV subclone pBLV913) (GenBank accession number EF600696) [37] with 102 reference sequences corresponding to known BLV genotypes. A phylogenetic tree was then constructed using the Kimura-2 model of nucleotide substitution with gamma distribution (K2+G) (Fig. 1). The ML phylogenetic tree showed congruent topologies, supported by moderate to high bootstrap values. As shown, the results were similar to those published in previous studies, in that the BLV strains were divided into nine genotypes [23–25, 29, 41] plus a new genotype-10, which was recently found only in Thailand [30]. Unexpectedly, all five Myanmar BLV strains grouped closely together with genotype-10 strains from Thailand.

Maximum likelihood (ML) phylogenetic tree based on partial BLV env-gp51 sequences from different geographical locations worldwide. A ML phylogenetic tree was constructed from partial (475 bp) BLV env-gp51 sequences from five Myanmar BLV strains and 102 sequences from known BLV strains representing the ten different BLV genotypes. These 102 nucleotide sequences were obtained from the GenBank nucleotide sequence database. The Myanmar BLV strains identified in this study are indicated by their sample ID and country name (Myanmar). Other isolates are indicated by accession number and country of origin. The Myanmar BLV strains identified in this study are marked as ●. Genotypes are indicated by numbers to the right of the figure. Genotype-10 (G-10) is highlighted in gray. The bar at the bottom of the figure denotes evolutionary distance
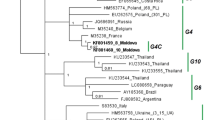
To clarify whether the Myanmar BLV strains belonged to genotype-10, we constructed a ML phylogenetic tree based on the complete (807 bp) env-gp51 sequences of the Myanmar strains and 98 references strains (corresponding to nucleotide positions 4925–5731 of the whole genome sequence of the FLK-BLV subclone pBLV913). Because strain p60 showed homology to L1, we used four out of the five strains for complete env-gp51 sequences analysis. As shown in Fig. 2, the phylogenetic tree was consistent with that for the partial env-gp51 sequences, i.e., the Myanmar BLV strains were classified as genotype-10.

Maximum likelihood (ML) phylogenetic tree showing complete BLV env-gp51 sequences from different geographical locations worldwide. The ML phylogenetic tree was constructed from complete (807 bp) BLV env-gp51 sequences from Myanmar BLV strains (submitted to the GenBank nucleotide sequence database and assigned accession numbers LC154064–LC154067) and 98 sequences from known BLV strains (representing ten different BLV genotypes from different locations). These reference sequences were obtained from the GenBank nucleotide sequence database. The Myanmar BLV strains identified in this study are indicated by sample ID and country name (Myanmar). Other isolates are indicated by accession number and country of origin. The Myanmar BLV strains identified in this study are marked as ●. Genotypes are indicated by numbers to the right of the figure. Genotype-10 (G-10) is highlighted in gray. The bar at the bottom of the figure denotes evolutionary distance
Nucleotide and amino acid sequence analyses of the partial env-gp51 region of BLV strains in Myanmar
Fourteen partial (475 bp) env-gp51 nucleotide sequences from the five Myanmar BLV strains and nine Thailand BLV strains were aligned with that of FLK-BLV subclone pBLV913 (Fig. 3A). The nucleotide sequence similarity of the 475 bp env-gp51 sequences from the five Myanmar BLV strains ranged from 98.15 % to 100 %. These five sequences were 93.08 % to 99.32 % similar to sequences corresponding to all ten genotypes. The Myanmar strains shared 13 nucleotide substitutions (nucleotides 317, 410, 431, 483, 525, 529, 552, 555, 561, 609, 615, 619, and 717) with the nine genotype-10 strains from Thailand and four nucleotide mutations (nucleotides 282, 405, 428, and 564) with some of them (Fig. 3A). Six other point mutations were also observed in the Myanmar strains.

Alignment of the nucleotide and deduced amino acid sequences of the partial BLV env-gp51 gene. Nucleotide sequences (A) and deduced amino acid sequences (B) of five Myanmar BLV strains were aligned with nine sequences of genotype-10 BLV strains from Thailand. The Myanmar BLV strains identified in this study are indicated by sample ID and country name (Myanmar) and are located in the lower part of the alignment. The Thailand isolates are indicated by accession number and country of origin. The genotype is indicated by the black bars at the far left of the figure. The numbers on the top of the sequences indicate the first and last nucleotide positions, and the positions of nucleotide substitutions. Numbers above the deduced amino acid sequence are amino acid residue numbers that indicate the start and end of each domain. The first, second, and third neutralizing domains (ND) as well as other epitopes are shown at the top of the alignment. Dots indicate identity with the FLK-BLV subclone pBLV913, which was used as the reference sequence
To gain further insight into the amino acid changes observed in the Myanmar BLV strains, we aligned the deduced amino acid sequences of the five partial env-gp51 sequences with representative genotype-10 strain sequences from Thailand. As shown in Fig. 3B, amino acid changes occurred mainly in the middle region of gp51, encompassing amino acid positions 89-246 of FLK-BLV subclone pBLV913. As expected, the Myanmar strains had five amino acid substitutions in common with the Thai BLV strains, and all were located in functional domains. For example, valine was replaced by alanine at residue 106 (V > A), which is located in both the first neutralizing domain (1st ND) and the CD4+ epitope region. Three amino acid substitutions, serine to phenylalanine at residue 137 (S > F), glutamine to arginine at residue 143 (Q > R), and isoleucine to threonine at residue 144 (I > T), were located in the 2nd ND. The last common amino acid substitution, proline to serine at residue 177 (P > S), was located in the CD8+ epitope and E epitope regions. In addition, three other mutations were observed in the Myanmar strains: Arginine was replaced by histidine at residue 121 (R > H), which is located in the G epitope; glutamine was replaced by arginine at residue 181 (Q > R) in the E epitope region, and glutamic acid was replaced by aspartic acid at residue 225 (E >D) in the 3rd ND.
ML phylogenetic tree based on BLV full genome sequences
To examine the complete genome sequence diversity of the Myanmar BLV strains, the complete BLV genome sequences of two Myanmar strains, L1 and S3, were obtained by clone-sequencing. The complete genome sequences showed highest similarity (96.06 %) with genotype-6 BLV strains (LC080657/Paraguay), while being only 94.66 % similar to genotype-2 BLV strains (FJ914764/Argentina).
A ML phylogenetic tree was constructed using sequences of Myanmar strains L1 and S3 and 27 previously reported BLV whole genome sequences (genotype-1, -2, -4, -6, and -9); the whole genome sequences of genotype-3, -5, -7, and -8 are unavailable (Fig. 4). The phylogenetic tree was consistent with the previous trees regarding the clustering of BLV genotypes, including genotype-1, -2, -4, -6, and -9 [29]. Interestingly, the two Myanmar BLV strains were located on separate branches (bootstrap values 100 % for every clade), confirming that genotype-10 is present in Myanmar.

Maximum likelihood (ML) phylogenetic tree constructed from complete BLV genomic sequences. The ML phylogenetic tree was constructed using the complete BLV genomic sequences from two Myanmar BLV strains (submitted to GenBank nucleotide sequence database and assigned accession numbers LC154848–LC154849), together with 27 reference sequences obtained from the GenBank nucleotide sequence database. One thousand replications were performed to calculate bootstrap values (indicated on the tree). The strains identified in this study are indicated by the sample ID and country name (Myanmar). Reference sequences are indicated by accession number and country of origin. Genotypes are indicated by numbers to the right of the figure. Genotype-10 (G-10) is highlighted in gray. The bar at the bottom of the figure denotes evolutionary distance
BLV genotype-10-specific mutations
To identify features that distinguish genotype-10 Myanmar BLV strains from previously known BLV strains belonging to genotype-1, -2, -4, -6, and -9, we aligned the deduced amino acid sequences of each BLV structural (gag, pro, pol, and env) and non-structural (tax, rex, R3, and G4) gene from two genotype-10 Myanmar BLV strains with that of 27 previously reported BLV whole genome sequences: the FLK-BLV subclone pBLV913 [37], strains LS1–LS3 from Uruguay [42], strains Arg41 and Arg38 from Argentina [43, 44], two strains from Japan [45] and the USA, and 16 strains from Peru, Paraguay, and Bolivia [29]. As summarized in Fig. 5, 22 unique amino acid substitutions were observed in Myanmar BLV strains L1 and S3. These included mutations (1) in the structural gene-encoded proteins: two substitutions (T38A and T366A) in Gag; one substitution (S52F) in Pro; four substitutions (V205L, I409V, P480S, and A826V) in Pol; and four substitutions (V106A, S137F, Q143R, and P177S) in Env; (2) in the regulatory proteins: six substitutions (N140K, V142E, I152T, D181N, E229D, and L273F) in Tax, three substitutions (S103F, L140P, and T156N) in Rex; and (3) in the accessory proteins: one substitution (K27N) in R3, and one substitution (L66P) in the G4 protein. We also demonstrated that each genotype harbored genotype-specific mutations that distinguished it from the others. These amino acid comparisons clearly showed that Myanmar BLV strains were significantly different from the other genotypes, further supporting the results generated by the full genome ML phylogenetic tree.

Summary of amino acid substitutions in the whole genome sequences of two Myanmar BLV strains when compared to 27 known BLV strains. Amino acid substitutions identified in at least two samples are shown. Genotypes are indicated by numbers to the far left of the figure. Each structural and regulatory gene-encoded glycoprotein is shown at the top of the figure. Numbers above the deduced amino acid sequence refer to the positions of amino acid substitutions. Genotype-10 (G-10) and genotype-10-specific mutations detected in both Myanmar BLV strains are highlighted in gray. Dots indicate identity with FLK-BLV subclone pBLV913, which was used as the reference
Discussion
We draw four major conclusions from the results of this study. The first relates to the distribution of BLV in Myanmar. The 66 samples collected from Myanmar revealed a relatively low level of BLV infection. Second, and most important, phylogenetic analysis based on partial and complete env-gp51 sequences revealed that Myanmar BLV strains clustered with genotype-10. Third, the ML phylogenetic tree based on complete BLV genome sequences clearly showed that Myanmar BLV strains separated from other known genotypes to form a genotype-10 clade, confirming the existence of this BLV genotype in Southeast Asia (Myanmar and Thailand). Fourth, a number of nucleotide and amino acid substitutions were found in the full BLV genome sequences and in the gp51 gene sequences of the Myanmar strains. Twenty-two BLV genotype-10-specific amino acid substitutions were observed in structural and non-structural genes from Myanmar BLV strains. Regarding the amino acid substitutions in the partial env-gp51 region, all were located in epitope regions (the CD4+ T cell epitope, 2nd ND, CD8+ T cell epitope, and G, B, and E epitope regions). Finally, most of the nucleotide substitutions were silent substitutions.
BLV is transmitted both horizontally and vertically [3, 46] via biological fluids, including infected cells in blood and milk [3, 35, 46, 47], as well as nasal secretions and saliva [36], and also by physical contact [35, 48] and sucking insects [49–51]. Iatrogenic routes include blood-contaminated dehorning devices [52, 53] and the common use of needles [53, 54] and plastic sleeves for rectal palpation [55], all of which make a substantial contribution to viral transmission. Regarding the presence of BLV in Myanmar cattle, multiple potential risk factors are associated with the BLV-positive results obtained in the current study. These include the breeding strategy used to develop draught and dairy cattle and to upgrade local breeds: cattle from most of the townships in Myanmar are crossed with many exotic breeds such as Holstein Frisian, Jersey, Shindi, and Thari [31]. Introduction of such breeds is highly associated with BLV infection in Japan [56] and other countries. Therefore, the introduction of infected cattle to Myanmar and the transfer of infected cattle from farm to farm within Myanmar, coupled with hybridizing or crossing of local breeds with imported breeds, might be a major driver of BLV infection in Myanmar [31]. In addition, embryo transfer technology is practiced in Myanmar [31]. Furthermore, special bulls are kept for breeding purposes in some areas of Myanmar; these bulls are part of a paid-for selective breeding program [31]. Thus, physical contact through breeding may be another route of transmission. Even though 9.1 % (6/66) of the samples examined herein were BLV-positive, the risk factors listed above lead us to hypothesize that BLV infection might be widespread in Myanmar, not only in the test areas, but also in other townships and territories.
Moratorio et al. (2010) [57] reported that phylogenetic analysis obtained using partial env-gp51 sequences was suitable for the quick assignment and establishment of phylogenetic relationships among BLV strains by comparing phylogenetic analysis obtained using partial or complete env-gp51 sequences. Indeed, our results are in agreement with this [57]. The phylogenetic analyses based on both partial and complete env-gp51 sequences support previous data indicating that BLV has ten genotypes [30]; we determined that the Myanmar BLV strains are genotype-10, along with the Thai strains. We thought it particularly interesting that the results of phylogenetic analyses based on partial or complete env-gp51 sequences clearly agreed with those based on phylogenetic analysis of whole BLV genome sequences from Myanmar strains. The phylogenetic results were confirmed by a ML phylogenetic tree based on the whole genome sequences of two novel Myanmar BLV strains and 27 previously reported strains (Fig. 4). The results were further confirmed by the discovery of BLV genotype-10-specific mutations within structural and non-structural BLV proteins (Fig. 5). Notably, the bootstrap value for the genotype-10 cluster was supported by a value of 100 in the ML phylogenetic tree constructed from whole BLV genome sequences. The genotype-10 Myanmar BLV strains showed a close genetic relationship; they are also related (albeit more distantly) to other previously reported genotypes.
Lee et al. suggest that BLV env-gp51 sequences derived from different geographical isolates show conservation of specific motifs depending on the genotype [27]. Our results support Lee et al. that even though nucleotide sequences show great variability (Fig. 3A), conservation of deduced amino acid sequences is strictly genotype dependent (Fig. 3B). Alignment of partial env-gp51 regions revealed a number of nucleotide and amino acid substitutions that were unique to genotype-10 (Fig. 3). All amino acid substitutions were located in functional epitopes or NDs, in agreement with previous molecular evolutionary analyses [12, 15, 41, 58]. Even though the biological functions of these substitutions are not clear, substitutions of S137F, Q143R and I144T were located in the 2nd ND which is involved in the interaction between gp51 and a receptor expressed on host cell membranes, suggesting that these substitutions could affect viral fusion and infectivity in vivo [13, 59]. In addition one mutation at residue 106 (V > A) was located in both the 1st ND and the CD4+ epitope, and one mutation at residue 177 (P > S) was located in the CD8+ and E epitope region. We found it interesting that the two Myanmar strains (S3 and S5/Myanmar) harbored a substitution at residue 121 (R > H) in the G epitope; this was previously described in genotype-4 BLV strains from Eastern Europe [25] and Chile [41, 57]. This epitope is both a conformational epitope and a target for MAbs that induce neutralization and inhibit syncytium formation [60, 61]. Likewise, two out of the 22 BLV genotype-10-specific amino acid substitutions were located in functional domains within structural and non-structural BLV proteins: aspartic acid to asparagine at residue 181 (D > N) in the leucine-rich domain [62] of Tax; and leucine to proline at residue 66 (L > P) within the arginine-rich nucleus region of G4 which is an RNA-binding domain [63].
As previously explained, Holstein, Frisian, Jersey, Shindi, and Thari breeds are imported to Myanmar to improve the local breeding stock [31]. Genotype-10 BLV strains were first detected in Thailand [30]. Therefore, we hypothesize that these exotic breeds might have been infected by BLV outside Myanmar and then subsequently transmitted the virus to cattle in Myanmar. This is supported by our detection of BLV in Friesian (sample ID: S1, S3, S5, L1, and L2) and Pyar Sein (p60) breeds. In addition five out of six BLV positive cattle were dairy cattle (sample ID: S1, S3, S5, L1, and L2), while Pyar Sein cattle (p60) was the only double-purpose breed (diary and drought cattle). As far as the farms and age of BLV positive cattle are concerned, cattle S1, S3 and S5 were 9-10 years old from the same farm, while L1 and L2 are 3-year old cattle from the same small-holding farm. Lastly, p60 was only the one positive cattle in another farm and was 2 years old. However, the exact geographical origin of Myanmar BLV genotype-10 strains remains uncertain. Further studies are needed to examine the diversity of BLV circulating in Myanmar, along with the BLV genotypes circulating in neighboring countries, including Laos, Bangladesh, Bhutan, India, and China. Since the samples tested in this study were collected from central Myanmar and their number was limited, we only detected genotype-10 strains. However, more genotypes might be detected if nationwide samples were obtained.
In conclusion, the present study provides evidence of the prevalence of BLV infection among cattle in Myanmar. Phylogenetic analysis demonstrated that BLV genotype-10 is present in Myanmar cattle. These results provide important information about the prevalence of BLV infection and will enable the implementation of appropriate cattle management policies and facilitate the development of more effective methods of BLV eradication.
References
Willems L, Burny A, Collete D, Dangoisse O, Dequiedt F, Gatot JS, Kerkhofs P, Lefebvre L, Merezak C, Peremans T, Portetelle D, Twizere JC, Kettmann R (2000) Genetic determinants of bovine leukemia virus pathogenesis. AIDS Res Hum Retroviruses 16:1787–1795
Aida Y, Murakami H, Takahashi M, Takeshima SN (2013) Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front Microbiol 4:328
Gillet N, Florins A, Boxus M, Burteau C, Nigro A, Vandermeers F, Balon H, Bouzar AB, Defoiche J, Burny A, Reichert M, Kettmann R, Willems L (2007) Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology 4:18
OIE (2009) World Animal Health information database-version: 1.4. World organisation for animal Health, Paris
Qualley DF, Boleratz BL (2014) Expression, purification, and characterization of full-length bovine leukemia virus Gag protein from bacterial culture. Protein Expr Purif 93:32–37
Willems L, Heremans H, Chen G, Portetelle D, Billiau A, Burny A, Kettmann R (1990) Cooperation between bovine leukaemia virus transactivator protein and Ha-ras oncogene product in cellular transformation. EMBO J 9:1577–1581
Felber BK, Derse D, Athanassopoulos A, Campbell M, Pavlakis GN (1989) Cross-activation of the Rex proteins of HTLV-I and BLV and of the Rev protein of HIV-1 and nonreciprocal interactions with their RNA responsive elements. New Biol 1:318–328
Willems L, Kerkhofs P, Dequiedt F, Portetelle D, Mammerickx M, Burny A, Kettmann R (1994) Attenuation of bovine leukemia virus by deletion of R3 and G4 open reading frames. Proc Natl Acad Sci USA 91:11532–11536
Florins A, Gillet N, Boxus M, Kerkhofs P, Kettmann R, Willems L (2007) Even attenuated bovine leukemia virus proviruses can be pathogenic in sheep. J Virol 81:10195–10200
Mamoun RZ, Astier T, Guillemain B, Duplan JF (1983) Bovine lymphosarcoma: expression of BLV-related proteins in cultured cells. J Gen Virol 64(Pt 9):1895–1905
Zarkik S, Decroly E, Wattiez R, Seidah NG, Burny A, Ruysschaert JM (1997) Comparative processing of bovine leukemia virus envelope glycoprotein gp72 by subtilisin/kexin-like mammalian convertases. FEBS Lett 406:205–210
Mamoun RZ, Morisson M, Rebeyrotte N, Busetta B, Couez D, Kettmann R, Hospital M, Guillemain B (1990) Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J Virol 64:4180–4188
Callebaut I, Voneche V, Mager A, Fumiere O, Krchnak V, Merza M, Zavada J, Mammerickx M, Burny A, Portetelle D (1993) Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. J Virol 67:5321–5327
Johnston ER, Radke K (2000) The SU and TM envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions. J Virol 74:2930–2935
Portetelle D, Couez D, Bruck C, Kettmann R, Mammerickx M, Van der Maaten M, Brasseur R, Burny A (1989) Antigenic variants of bovine leukemia virus (BLV) are defined by amino acid substitutions in the NH2 part of the envelope glycoprotein gp51. Virology 169:27–33
Bruck C, Mathot S, Portetelle D, Berte C, Franssen JD, Herion P, Burny A (1982) Monoclonal antibodies define eight independent antigenic regions on the bovine leukemia virus (BLV) envelope glycoprotein gp51. Virology 122:342–352
Camargos MF, Stancek D, Rocha MA, Lessa LM, Reis JK, Leite RC (2002) Partial sequencing of env gene of bovine leukaemia virus from Brazilian samples and phylogenetic analysis. J Vet Med B Infect Dis Vet Public Health 49:325–331
Coulston J, Naif H, Brandon R, Kumar S, Khan S, Daniel RC, Lavin MF (1990) Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: comparison with other isolates. J Gen Virol 71(Pt 8):1737–1746
Beier D, Blankenstein P, Marquardt O, Kuzmak J (2001) Identification of different BLV provirus isolates by PCR, RFLPA and DNA sequencing. Berl Munch Tierarztl Wochenschr 114:252–256
Felmer R, Munoz G, Zuniga J, Recabal M (2005) Molecular analysis of a 444 bp fragment of the bovine leukaemia virus gp51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet Microbiol 108:39–47
Camargos MF, Pereda A, Stancek D, Rocha MA, dos Reis JK, Greiser-Wilke I, Leite RC (2007) Molecular characterization of the env gene from Brazilian field isolates of Bovine leukemia virus. Virus Genes 34:343–350
Monti G, Schrijver R, Beier D (2005) Genetic diversity and spread of Bovine leukaemia virus isolates in Argentine dairy cattle. Arch Virol 150:443–458
Matsumura K, Inoue E, Osawa Y, Okazaki K (2011) Molecular epidemiology of bovine leukemia virus associated with enzootic bovine leukosis in Japan. Virus Res 155:343–348
Balic D, Lojkic I, Periskic M, Bedekovic T, Jungic A, Lemo N, Roic B, Cac Z, Barbic L, Madic J (2012) Identification of a new genotype of bovine leukemia virus. Arch Virol 157:1281–1290
Rola-Luszczak M, Pluta A, Olech M, Donnik I, Petropavlovskiy M, Gerilovych A, Vinogradova I, Choudhury B, Kuzmak J (2013) The molecular characterization of bovine leukaemia virus isolates from Eastern Europe and Siberia and its impact on phylogeny. PLoS One 8:e58705
Polat M, Ohno A, Takeshima SN, Kim J, Kikuya M, Matsumoto Y, Mingala CN, Onuma M, Aida Y (2015) Detection and molecular characterization of bovine leukemia virus in Philippine cattle. Arch Virol 160:285–296
Lee E, Kim EJ, Joung HK, Kim BH, Song JY, Cho IS, Lee KK, Shin YK (2015) Sequencing and phylogenetic analysis of the gp51 gene from Korean bovine leukemia virus isolates. Virol J 12:64
Ochirkhuu N, Konnai S, Odbileg R, Nishimori A, Okagawa T, Murata S, Ohashi K (2016) Detection of bovine leukemia virus and identification of its genotype in Mongolian cattle. Arch Virol 161:985–991
Polat M, Takeshima SN, Hosomichi K, Kim J, Miyasaka T, Yamada K, Arainga M, Murakami T, Matsumoto Y, de la Barra Diaz V, Panei CJ, Gonzalez ET, Kanemaki M, Onuma M, Giovambattista G, Aida Y (2016) A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology 13:4
Lee E, Kim EJ, Ratthanophart J, Vitoonpong R, Kim BH, Cho IS, Song JY, Lee KK, Shin YK (2016) Molecular epidemiological and serological studies of bovine leukemia virus (BLV) infection in Thailand cattle. Infect Genet Evol 41:245–254
National Consultative Committee M (2013) National report on animal genetic resources, The Union of Myanmar. National Consultative committee, Myanmar
Licursi M, Inoshima Y, Wu D, Yokoyama T, Gonzalez ET, Sentsui H (2003) Provirus variants of bovine leukemia virus in naturally infected cattle from Argentina and Japan. Vet Microbiol 96:17–23
Hughes SH, Shank PR, Spector DH, Kung HJ, Bishop JM, Varmus HE, Vogt PK, Breitman ML (1978) Proviruses of avian-sarcoma virus are terminally redundant, co-extensive with unintegrated linear DNA and integrated at many sites. Cell 15:1397–1410
Takeshima SN, Kitamura-Muramatsu Y, Yuan Y, Polat M, Saito S, Aida Y (2015) BLV-CoCoMo-qPCR-2: improvements to the BLV-CoCoMo-qPCR assay for bovine leukemia virus by reducing primer degeneracy and constructing an optimal standard curve. Arch Virol 160:1325–1332
Ohno A, Takeshima SN, Matsumoto Y, Aida Y (2015) Risk factors associated with increased bovine leukemia virus proviral load in infected cattle in Japan from 2012 to 2014. Virus Res 210:283–290
Yuan Y, Kitamura-Muramatsu Y, Saito S, Ishizaki H, Nakano M, Haga S, Matoba K, Ohno A, Murakami H, Takeshima SN, Aida Y (2015) Detection of the BLV provirus from nasal secretion and saliva samples using BLV-CoCoMo-qPCR-2: Comparison with blood samples from the same cattle. Virus Res 210:248–254
Rovnak J, Boyd AL, Casey JW, Gonda MA, Jensen WA, Cockerell GL (1993) Pathogenicity of molecularly cloned bovine leukemia virus. J Virol 67:7096–7105
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Rodriguez SM, Golemba MD, Campos RH, Trono K, Jones LR (2009) Bovine leukemia virus can be classified into seven genotypes: evidence for the existence of two novel clades. J Gen Virol 90:2788–2797
Wang H, Norris KM, Mansky LM (2003) Involvement of the matrix and nucleocapsid domains of the bovine leukemia virus Gag polyprotein precursor in viral RNA packaging. J Virol 77:9431–9438
Dube S, Dolcini G, Abbott L, Mehta S, Dube D, Gutierrez S, Ceriani C, Esteban E, Ferrer J, Poiesz B (2000) The complete genomic sequence of a BLV strain from a Holstein cow from Argentina. Virology 277:379–386
Dube S, Abbott L, Dube DK, Dolcini G, Gutierrez S, Ceriani C, Juliarena M, Ferrer J, Perzova R, Poiesz BJ (2009) The complete genomic sequence of an in vivo low replicating BLV strain. Virol J 6:120
Sagata N, Yasunaga T, Ohishi K, Tsuzuku-Kawamura J, Onuma M, Ikawa Y (1984) Comparison of the entire genomes of bovine leukemia virus and human T-cell leukemia virus and characterization of their unidentified open reading frames. EMBO J 3:3231–3237
Rodriguez SM, Florins A, Gillet N, de Brogniez A, Sanchez-Alcaraz MT, Boxus M, Boulanger F, Gutierrez G, Trono K, Alvarez I, Vagnoni L, Willems L (2011) Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Viruses 3:1210–1248
Ferrer JF, Marshak RR, Abt DA, Kenyon SJ (1978) Persistent lymphocytosis in cattle: its cause, nature and relation to lymphosarcoma. Ann Rech Vet 9:851–857
Kono Y, Sentsui H, Arai K, Ishida H, Irishio W (1983) Contact transmission of bovine leukemia virus under insect-free conditions. Nihon Juigaku Zasshi 45:799–802
Bech-Nielsen S, Piper CE, Ferrer JF (1978) Natural mode of transmission of the bovine leukemia virus: role of bloodsucking insects. Am J Vet Res 39:1089–1092
Ferrer JF, Piper CE (1981) Role of colostrum and milk in the natural transmission of the bovine leukemia virus. Cancer Res 41:4906–4909
Ohshima K, Okada K, Numakunai S, Yoneyama Y, Sato S, Takahashi K (1981) Evidence on horizontal transmission of bovine leukemia virus due to blood-sucking tabanid flies. Nihon Juigaku Zasshi 43:79–81
DiGiacomo RF, Darlington RL, Evermann JF (1985) Natural transmission of bovine leukemia virus in dairy calves by dehorning. Can J Comp Med 49:340–342
Lassauzet ML, Thurmond MC, Johnson WO, Stevens F, Picanso JP (1990) Effect of brucellosis vaccination and dehorning on transmission of bovine leukemia virus in heifers on a California dairy. Can J Vet Res 54:184–189
Hopkins SG, Evermann JF, DiGiacomo RF, Parish SM, Ferrer JF, Smith S, Bangert RL (1988) Experimental transmission of bovine leukosis virus by simulated rectal palpation. Vet Rec 122:389–391
Van der Maaten MJ, Miller JM, Schmerr MJ (1981) In utero transmission of bovine leukemia virus. Am J Vet Res 42:1052–1054
Kobayashi S, Hidano A, Tsutsui T, Yamamoto T, Hayama Y, Nishida T, Muroga N, Konishi M, Kameyama K, Murakami K (2014) Analysis of risk factors associated with bovine leukemia virus seropositivity within dairy and beef breeding farms in Japan: a nationwide survey. Res Vet Sci 96:47–53
Moratorio G, Obal G, Dubra A, Correa A, Bianchi S, Buschiazzo A, Cristina J, Pritsch O (2010) Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch Virol 155:481–489
Zhao X, Buehring GC (2007) Natural genetic variations in bovine leukemia virus envelope gene: possible effects of selection and escape. Virology 366:150–165
Gatot JS, Callebaut I, Van Lint C, Demonte D, Kerkhofs P, Portetelle D, Burny A, Willems L, Kettmann R (2002) Bovine leukemia virus SU protein interacts with zinc, and mutations within two interacting regions differently affect viral fusion and infectivity in vivo. J Virol 76:7956–7967
Bruck C, Portetelle D, Burny A, Zavada J (1982) Topographical analysis by monoclonal antibodies of BLV-gp51 epitopes involved in viral functions. Virology 122:353–362
Bruck C, Portetelle D, Mammerickx M, Mathot S, Burny A (1984) Epitopes of bovine leukemia virus glycoprotein gp51 recognized by sera of infected cattle and sheep. Leuk Res 8:315–321
Willems L, Kettmann R, Burny A (1991) The amino acid (157-197) peptide segment of bovine leukemia virus p34tax encompass a leucine-rich globally neutral activation domain. Oncogene 6:159–163
Alexandersen S, Carpenter S, Christensen J, Storgaard T, Viuff B, Wannemuehler Y, Belousov J, Roth JA (1993) Identification of alternatively spliced mRNAs encoding potential new regulatory proteins in cattle infected with bovine leukemia virus. J Virol 67:39–52
Acknowledgments
We thank LBVD members and farm owners of Lewe, Pyinmana and Shwe Myo Livestock Zone for assistance with sampling the farms in Myanmar. We are grateful to the Support Unit at the Bio-material Analysis, RIKEN BSI Research Resources Center, for help with sequence analysis. This work was supported by Grants-in-Aid for Scientific Research A (No: 18255013) from the Japan Society for the Promotion of Science, and by a Grant from Integration Research for Agriculture and Interdisciplinary Fields in Japan (No.: 14538311).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Animal handling and research ethics
All animals were handled by veterinarians from the University of Veterinary Science in Yezin Myanmar in strict accordance with good animal practice following the guidelines of the University of Veterinary Science. This experiment is approved by the Committee on the Ethics of Animals for Research at the Faculty of Agriculture, Kagoshima University (Certificate No: H28 NOU 005).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Polat, M., Moe, H.H., Shimogiri, T. et al. The molecular epidemiological study of bovine leukemia virus infection in Myanmar cattle. Arch Virol 162, 425–437 (2017). https://doi.org/10.1007/s00705-016-3118-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3118-y