
Abstract
Bovine group A rotavirus (bovine RVA) is recognized as a major cause of severe gastroenteritis in newborn calves. The purpose of this study was to estimate the prevalence and identify the genotypes of circulating bovine RVA in newborn diarrheic calves. Two hundred fifty-three stool samples of diarrheic calves up to 1 month old were collected from 42 industrial dairy farms in two Iranian provinces during March 2010 to February 2012. All collected samples were screened for the presence of bovine RVA by RT-PCR, and the G and P genotypes were determined by semi-nested multiplex RT-PCR assay. The results of RT-PCR indicated that 49.4 % (125 out of 253) of the samples were positive for bovine RVA. The G and P genotyping of a subset of positive samples (n = 85) by semi-nested multiplex RT-PCR revealed that G6 (55.3 %) and G10 (43.5 %) and P[5] (51.8 %) and P[11] (27 %) were the most prevalent G and P genotypes, respectively. G6P[5] was the dominant genotype (35.3 %), followed by G10P[5], G10P[11] and G6P[11], with prevalence rates of 16.5 %, 15.3 % and 10.6 %, respectively. Sequence analysis of 20 VP7 and four VP4 genes showed highest nucleotide sequence identity with the corresponding genes of strains RVA/Cow-tc/GBR/UK/1973/G6P7[5] and RVA/Cow-tc/USA/B223/XXXX/G10P[11]. The results of this study reveal the diversity of G and P genotypes in bovine RVA samples from diarrheic Iranian calves and expands our knowledge of bovine RVA infections in the Middle East. These results also highlight the importance of producing of an effective rotavirus vaccine and its inclusion in the national cattle immunization program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bovine group A rotavirus (bovine RVA) is recognized as the most common cause of severe gastroenteritis in cattle, resulting in significant morbidity and mortality in newborn calves and economic losses. According to numerous reports, this viral infection has had a negative impact on the cattle industry worldwide [14, 31]. Diarrhea in neonatal calves caused by bovine RVA has been reported in both dairy and beef cattle herds [31]. The worldwide prevalence rate for BRV infection has been reported to range from 7 % to 94 %, while some studies have reported moderate prevalence rates of approximately 30–40 % [18, 59, 64].
Bovine RVA belongs to the genus Rotavirus in the family Reoviridae. The genome of this virus consists of 11 segments of double-stranded RNA [10, 17] surrounded by a triple-layered capsid. The rotavirus genome encodes six structural (VP1–VP4, VP6, and VP7) and six nonstructural (NSP1-NSP6) proteins [17]. Variation in VP6, the protein forming the middle layer of the virus capsid, is used to define eight major rotavirus groups, which are alphabetically named A-H [44]. Rotavirus group A is considered the most important group because of its high prevalence and pathogenicity in humans and a variety of animals [43].
Classification of rotaviruses in group A is based on neutralizing antibodies against the capsid proteins VP7 and VP4, resulting in a dual genotyping system in which G (glycoprotein) and P (protease-sensitive protein) genotypes, respectively, are defined. To date, at least 12 different G genotypes (G1-G3, G5, G6, G8, G10, G11, G15, G17, G21, G24) and 11 different P genotypes (P[1], P[3], P[5]-P[7], P[11], P[14], P[17], P[21], P[29], P[38]) have been reported in cattle, with G6, G8, G10, P[1], P[5] and P[11] being the most common genotypes identified worldwide [52]. During bovine RVA epidemiological studies, G6 and G10 genotypes have frequently been detected in different countries, including Turkey, India, Thailand, France, Tunisia, Denmark, Hungary, Canada, the United States, Argentina, Australia and New Zealand [4, 22, 30, 32, 46, 59, 62, 66]. Compared to G6 and G10 strains, the overall frequency of G8 strains is much lower; however, it appears to be important in some countries. This genotype was reported to be the dominant strain in Japan and Tunisia during 1995–1996 and 2001–2002, respectively [19, 20].
Several studies have been conducted using ELISA to determine the prevalence of bovine RVA in Iranian cattle from different provinces, including Fars, Tehran, Chahar Mahal Bakhtiari, Khorasan and West Azarbayjan [1, 7, 37, 40, 45, 49]. However, no published study has estimated the prevalence of RVA using the more reliable and sensitive RT-PCR. More recently, Madadgar and colleagues, who studied the prevalence of bovine RVA in Iranian calves by ELISA, reported a prevalence of 28.2 % [40]. Furthermore, a few studies on G genotyping and only one study on P genotyping have been done on bovine RVA in Iran [40, 45]. Thus, the objective of this study was to determine the prevalence of bovine rotavirus A by RT-PCR in fecal samples collected from diarrheic calves. Stool specimens of the diarrheic calves from prominent dairy farms located in the provinces of Tehran and Alborz were collected during two years from spring 2010 to winter 2012 and analyzed by RT-PCR targeting the VP6 gene. To get more insights into the molecular epidemiology and genetic relationship of the virus to the prevailing genotypes worldwide, a subset of bovine RVA-positive samples were genotyped (G and P), and a few were then further analyzed by partial genome sequencing.
Materials and methods
Samples
A total of 253 fecal samples from diarrheic calves (Bos taurus) up to one month old were collected from 42 industrial dairy farms in two provinces (Tehran and Alborz) in Iran. Selection of farms and the number of samples collected from the animals of each farm (based on the population of each farm) were computed in each season by GIS (Geographical Information System) software of the Iranian Veterinary Organization. Samples were collected during all seasons for a two-year period starting from March 2010 to February 2012. Seventy-two samples were collected during the first year of study, while 181 samples were collected in the second year of study. It is worth mentioning that the number of diarrheic samples collected per outbreak for each farm was related to the size of the herd (one calf sampled per 50 calves). Once collected, the samples were immediately stored at -40 °C until analysis.
Nucleic acid extraction
Viral RNA was extracted from 20 % fecal suspensions in phosphate-buffered saline using the NucliSENS® EasyMAG™ platform (bioMérieux, Marcy L’Etoile, France) according to the manufacturer’s instructions and was then stored at -40 °C.
Bovine RVA detection, genotyping and sequencing
Bovine RVA was detected by endpoint RT-PCR, using a OneStep RT-PCR Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. A VP6-F/VP6-R primer set [34] was used to detect the VP6 gene of bovine RVA (Table 1).
A subset of rotavirus-positive samples selected from different farms and different seasons were G- and P-genotyped. Samples for genotyping were selected in order to get one representative positive sample genotyped per episode of diarrhea per farm and per year of surveillance. Rotavirus genotyping was performed by semi-nested multiplex RT-PCR according to the EuroRotaNet protocol (http://www.eurota.net/docs.php). A set of previously described primers [26, 27] were used to amplify five G genotypes (G5, G6, G8, G10 and G11) and five P genotypes (P[1], P[5] - P[7] and P[11]) most commonly found in cattle.
Finally, in order to get more insights about the strains of rotaviruses circulating in the country, representative strains from each detected genotype (G6: n = 19, G10: n = 6, P[5]: n = 4, P[11]: n = 2) were randomly selected, and their VP7 or VP4 partial coding genes were sequenced.
All of the sequencing reactions were performed using an ABI PRISM® Big Dye® Terminator Cycle Sequencing Kit on a 3130XL DNA Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Sequence analysis
Molecular Evolutionary Genetics Analysis (MEGA) software v5 [60] was used to calculate sequence identities between strains using the pairwise distance (p-distance) substitution model. Phylogenetic analysis was performed online on the Phylogeny.fr platform [13] using the maximum-likelihood method. For these analyses, sequences of reference strains available in the National Center for Biotechnology Information GenBank database (http://www.ncbi.nlm.nih.gov/genbank/) were used.
Results
Prevalence of bovine RVA in cattle
Among the 253 samples collected during the entire two-year study, bovine RVA was detected in 125 samples (49.4 %). Compared to the bovine RVA detection rate in the first year (59.7 %), the prevalence rate was significantly lower (45.3 %) during the second year of the study. Although bovine RVA was detected all year round, the detection rate fluctuated according to the season, with a peak of infections during the cold seasons including winter and autumn (Table 2).
G and P genotypes of bovine RVA
Among the 125 rotavirus-positive samples, rotavirus G and P genotyping was performed on a subset of 85 samples only. G genotyping revealed that G6 was the most prevalent G genotype (55.3 %), followed by G10 (43.5 %). Co-infection of G6 and G10 genotypes were detected at a low prevalence (1.2 %), whereas genotypes G5, G8 and G11 were not detected in the samples (Table 3). P genotyping indicated that P[5] was the dominant P genotype, with a prevalence of 51.8 %, followed by P[11] (27 %). Coinfection with genotypes P[5] and P[11] was not detected in any of the samples, while 21.2 % of the samples were not of the P genotype (Table 3). P[1], P[6] and P[7] genotypes were not detected among the Iranian calves in this study.
G6P[5] was the predominant genotype, with a prevalence rate of 35.3 %, followed by G10P[5] (16.5 %), G10P[11] (15.3 %), and G10P-undetermined strains (11.8 %). Other combinations of G and P genotypes were also detected (i.e. G6P[11], G6P-undetermined, and coinfections), but with a prevalence of lower than 10 % (Table 3).
Rotavirus sequence analysis
The results of sequencing of the selected samples confirmed the genotyping results obtained by RT-PCR. According to MEGA5 p-distance studies, sequence analysis of the selected G6 RT-PCR-positive samples confirmed the presence of G6 genotypes among these samples. The 19 Iranian G6 sequences showed a high level of nucleotide (86.5 % to 97.0 %) and amino acid sequence identity (91 % to 100 %) to each other. These G6 samples appeared to be more closely related to the RVA/Cow-tc/GBR/UK/1973/G6P7[5] strain (nucleotide sequence identity 81.2 %-94.9 %; amino acid sequence identity 91 %-98 %) compared to the RVA/Cow-tc/USA/B223/XXXX/G10P[11] strain (nucleotide and amino acid sequence identity 72.9 %-74.4 % and 79.6 %-83.1 %, respectively). Furthermore, sequence analyses of the selected G10 RT-PCR-positive samples confirmed the presence of G10 genotypes among these samples. All six G10 sample sequences exhibited a high level of nucleotide (96.9 %-100 %) and amino acid (98.6 %-100 %) sequence similarity to each other and were most closely related to the RVA/Cow-tc/USA/B223/XXXX/G10P[11] strain (nucleotide and amino acid sequence identity 94.7 %-95.4 % and 96.3 %-96.4 %, respectively (Table 4).
Sequence analyses of the selected P[5] RT-PCR-positive samples also confirmed the presence of P[5] genotypes in these samples. All four P[5] sample sequences revealed a high level of nucleotide and amino acid sequence identity (97.2 %-99 % and 97.8 %-100 %, respectively) to each other, and were more closely related to the RVA/Cow-tc/GBR/UK/1973/G6P7[5] strain (nucleotide sequence identity, 86.6 %-89.7 %; amino acid sequence identity, 88.2 %-92.3 %) than compared to the RVA/Cow-tc/USA/B223/XXXX/G10P[11] strain (nucleotide sequence identity, 47.5 %-48 %; amino acid sequence identity, 50.2 %-54.7 %). Sequence analysis of the two selected P[11] RT-PCR-positive samples confirmed the presence of the P[11] genotype and showed that they are most closely related to the RVA/Cow-tc/USA/B223/XXXX/G10P[11] strain (nucleotide and amino acid sequence identity, 97.7 %-98 and 98.2 %, respectively) (Table 5).
Sequences of the Iranian bovine RVA strains identified in this study were submitted to the GenBank database under the following accession numbers: KM609892 to KM609911 and KX517906 to KX517910 for VP7 genes and KM609912 to KM609915 and KX517911 to KX517912 for VP4 genes.
Phylogenetic analysis
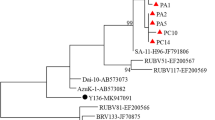
The results of phylogenic analysis of a portion of the VP7 coding gene of the 19 G6 and six G10 sequenced in this study are illustrated in Fig. 1 and Fig. 2. The relationship of these genes to other G6 and G10 strains detected in humans and cattle is also shown. According to the phylogenetic analysis of both nucleotide and the deduced amino acid sequences, the Iranian G6 isolates are grouped in two distinct small clusters and are related to the UK strain. Also, as highlighted in Fig. 1 and Fig. 2, all of the Iranian bovine G10 strains in this study formed a single cluster with the B223 strain.

Phylogenetic tree of partial VP7 coding gene sequences of bovine G6 and G10 rotaviruses. The tree was generated by Phylogeny.fr using the maximum-likelihood method implemented in the PhyML programme. Bootstrap values were calculated for 1000 replicates and are indicated at each node when ≥70 %. For each isolate, the GenBank accession number is shown, followed by the full rotavirus strain name, as recommended by the Rotavirus Classification Working Group (RCWG) [42]. Isolates from this study are underlined. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site

Phylogenetic tree based on partial VP7 amino acid sequences deduced from partial VP7 coding gene sequences of bovine G6 and G10 rotaviruses. The tree was generated by Phylogeny.fr using the maximum-likelihood method implemented in the PhyML programme. Bootstrap values were calculated for 1000 replicates and are indicated at each node when ≥70 %. For each isolate, the GenBank accession number is shown, followed by the full rotavirus strain name, as recommended by the Rotavirus Classification Working Group (RCWG) [42]. Isolates from this study are underlined. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site
As illustrated in Fig. 3 and Fig. 4, all of the Iranian bovine P[5] strains in this study exhibited close similarity to other P[5] strains detected in cattle and formed one cluster with the UK strain. Moreover, the Iranian bovine P[11] isolates (G10P[11] and G6P[11]) exhibited close similarity to other P[11] strains detected in humans, cattle and camels and formed a single cluster with the B223 strain.

Phylogenetic tree of partial VP4 coding gene sequences of bovine P[5] and P[11] rotaviruses. The tree was generated by Phylogeny.fr using the maximum-likelihood method implemented in the PhyML programme. Bootstrap values were calculated for 1000 replicates and are indicated at each node when ≥70 %. For each isolate, the GenBank accession number is shown, followed by the full rotavirus strain name, as recommended by the Rotavirus Classification Working Group (RCWG) [42]. Isolates from this study are underlined. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site

Phylogenetic tree of VP4 partial amino acid deduced from partial VP4 coding gene sequences of bovine P[5] and P[11] rotaviruses. The tree was generated by Phylogeny.fr using the maximum-likelihood method implemented in the PhyML programme. Bootstrap values were calculated for 1000 replicates and are indicated at each node when ≥70 %. For each isolate, the GenBank accession number is shown, followed by the full rotavirus strain name, as recommended by the Rotavirus Classification Working Group (RCWG) [42]. Isolates for this study are underlined. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site
Discussion
According to previous studies from different regions of the world, the prevalence of rotavirus in fecal samples of calves with diarrhea ranges from moderate to very high (30 % to 94 %) [5, 14, 21, 24, 45]. In Iran, several studies have shown that the prevalence of rotavirus infection in the diarrheic calves ranges from 26.9 % to 34 %, depending on the season and province studied [1, 7, 37, 40, 45, 49]. All of the previously conducted studies on bovine RVA in Iran were based on the ELISA method. However, in the present study, analyzing samples from Tehran and Alborz provinces, a significantly higher prevalence rate (49.4 %) was obtained by using the more sophisticated RT-PCR technique. This technique is significantly more sensitive than ELISA assays [28, 68], and this may explain the higher rotavirus prevalence rate observed in our studies.
In the two-year study, bovine RVA was detected in all seasons throughout the year, with a peak during cold temperatures in the winters. This high incidence rate during the winters could be explained by the effect of temperature, humidity and precipitation [38, 69]. Similar results have been reported previously, showing a higher rate of rotavirus infection in the cooler season, with most cases occurring between November and April [29, 56]. However, there are exceptions, such as in Egypt, where rotavirus infections have been reported to peak from July to November [50, 67]. There is no study on seasonal prevalence of bovine rotaviruses in Iran, although a few studies in Iran have shown the winter season to be the peak season for human rotavirus gastroenteritis [16, 38, 47, 69].
Worldwide studies have shown G6 and G10 with either P[5] or P[11] to be the most common genotypes within the calf population, while G8 and P[1] genotypes are less common [2, 55, 59]. In the present study, among the G5, G6, G8, G10 and G11 genotypes, only G6 and G10 genotypes were found in the collected stool samples. Furthermore, our results demonstrate that the distribution of these genotypes follows a worldwide trend: the G6 genotype was the most frequently identified in this study, occurring in 55.3 % of rotavirus infections, and the G10 genotype, with a slight difference, was the second most prevalent G genotype (43.5 %). The results of this study are consistent with those of a study conducted by Mayameei and co-workers in Iran. Those researchers reported the G6 genotype to be the most prevalent genotype (48 %) in Tehran and its surrounding area [45]. However, these genotyping results are different than those reported by Madadgar et al., which could be due to differences in the duration of the studies and the loss of seasonal selection in the latter study [40].
In this study, we also determined the P genotypes of bovine RVA circulating in Iran. P[1], P[5], P[6], P[7] and P[11] were detected in the studied samples, with the P[5] genotype being the most prevalent P genotype (51.8 %), followed by P[11] (27 %). Our results for P genotyping showed the same pattern of prevalence reported from many countries including Japan, Latin America, the United States, Australia and most of the European countries [2, 5, 10, 22, 33, 55, 59]. However, a few reports from India, Italy, Mexico and Turkey have shown P[11] to be the most frequent P genotype [4, 18, 48, 65]. Here, we report that the P genotype pattern in Iran did not follow the pattern of its western neighbor country, Turkey, nor the pattern of India to the east. It should be noted that in 21.8 % of the samples, we were unable to detect the P genotype, which might be due to the sensitivity of genotyping RT-PCR, considering primer mismatches at binding sites due to the accumulation of single-nucleotide polymorphisms. Alternatively, it is possible that these samples had other P genotypes that are not included in the multiplex RT-PCR assay used in this study. Unfortunately, our attempts to genotype these samples by amplifying and sequencing the VP4 coding gene using well-established VP4 consensus primers (i.e.; the Con3/Con2 and VP4-F/VP4-R primer sets) remained unsuccessful (data not shown) [23, 57].
The present study revealed that G6P[5] was the predominant bovine RVA genotype in Iran. This observation is consistent with studies from the USA, South America, Japan, and Europe, where G6P[5] was reported to be the most prevalent genotype in calves with diarrhea [18, 22, 33, 39]. This genetic combination was also reported to be the most prevalent combination in studies from Australia, Argentina, Brazil, the UK, Ireland, Japan and the United States [2, 4, 5, 10, 11, 33, 36, 46, 55, 58, 59]. Bovine RVA mixed infections occurred at low frequency during this study (1.2 %), as observed in several studies showing multiple G and/or P genotypes within a single stool sample [8, 18, 20, 22, 33, 58, 59]. Mixed infections are usually rare [8, 19, 20, 22, 33]. However, Swiatec and his colleagues reported mixed infections in 30.8 % of studied cases [59]. The occurrence of mixed infections makes natural reassortment between strains feasible and could result in natural diversity of rotaviruses [54].
Phylogenic studies of both partial VP7 genes and their deduced amino acid sequences of the G6 and G10 genotypes in this study demonstrated a relationship to the UK and B223 strains as well as to other human and bovine G6 and G10 genotypes reported from other countries. More-detailed examination of the phylogenic trees revealed that the Iranian G6 strains could be grouped into two small clusters. One of the clusters includes eight G6 strains together with those from Tunisia, Hungary, Argentina and Iran, and also with a human G6 strain from France [6, 30, 40]. The other cluster included seven Iranian G6 strains together with the UK, NCDV, and WC3 bovine strains and other bovine strains from the UK, the USA, Canada, China, Denmark, Japan, Australia and Iran [15, 40, 46, 59, 66]. Also, phylogenic analysis of translated sequences tended to give the same clustering and sub-clustering results, which confirms the prevalence of two partially distinct G6 bovine RVA strains among Iranian calves. The Iranian bovine G10 strains identified in this study appeared to be closely related to the B223 strain and other strains from Turkey, India, China, Thailand, Australia, Argentina, Canada, the USA and Iran [4, 6, 12, 40, 59, 61, 66]. A phylogenic analysis based on the deduced amino acids sequence of a portion of the coding region of the VP7 gene of G10 strains yielded the same clustering obtained using nucleotide sequences.
The results of phylogenetic analysis of P[5] and P[10] in this study revealed similarity of the Iranian strains to strains from Asia, Europe, Africa, North and South America and Australia. The Iranian bovine P[5] strains of this study exhibited close similarity to other P[5] strains detected in cattle, and formed a single cluster with the UK strain and other bovine strains from Turkey, Korea, Japan, Thailand, Denmark, Argentina, the USA and Iran [4, 6, 12, 40, 41, 46, 53, 59, 61, 66], and also a human G1P[5] strain of CJN-M from the USA [51]. Moreover, phylogenic analysis based on nucleotide and deduced amino acid sequences indicated that the Iranian bovine P[11] isolate (G10P[11]) exhibited close similarity to other P[11] strains detected in humans, cattle and camels and were grouped in one cluster with the well-known B223 strain and other strains from Turkey, India, China, Thailand, Ireland, Sudan, Australia, Argentina, the USA and Iran [4, 6, 12, 35, 40, 46, 59, 61, 66].
In conclusion, this study demonstrates the high prevalence of bovine group A rotaviruses in Iranian cattle, with almost half of the diarrheic calves shedding this virus. The most frequent bovine RVA strains were a combination of G6 and G10 with P[5] and P[11] genotypes. This study provides further information regarding rotavirus P genotypes and the sequence analysis of viruses found in Iranian cattle. Moreover, because these specimens were obtained from the largest husbandry centers in Iran, located in the provinces of Tehran and Alborz, these data might be valuable for emphasizing the importance of producing an effective rotavirus vaccine and its inclusion in the national cattle immunization program. As yet, no vaccine against bovine RVA is being produced in Iran. However, many commercially available bovine RVA vaccines are based on G6 and G10 strains of bovine RVA [8, 63] and thus seem to be suitable for the prevention of severe rotavirus infections in Iranian cattle. For example, the Kolibin RC Neo vaccine [9] is being used on a few Iranian farms, providing satisfactory immunity to the vaccinated herds [3]. This study could be considered for making any decision about choosing an international vaccine and/or for developing an effective national rotavirus vaccine for Iranian cattle.
References
Afshari Safavi EA, Mohammadi GR, Rad M, Naghibi A (2012) A case-ontrol study of association between diarrhea in newborn calves and infection with rotavirus and coronavirus in some industrial dairy herds of Mashhad area, Iran in 2008. Arch Razi Inst 67:35–41
Alfieri AF, Alfieri AA, Barreiros MA, Leite JP, Richtzenhain LJ (2004) G and P genotypes of group A rotavirus strains circulating in calves in Brazil, 1996–1999. Vet Microbiol 99:167–173
Alimolaei M (2012) Veterinary vaccines and their method of administration. Iranian Veterinarians Website, Iran. http://www.mainvets.com/vaccine-section/animalvacciness/1032-vaccine6.html
Alkan F, Ozkul A, Oguzoglu TC, Timurkan MO, Caliskan E, Martella V, Burgu I (2010) Distribution of G (VP7) and P (VP4) genotypes of group A bovine rotaviruses from Turkish calves with diarrhea, 1997–2008. Vet Microbiol 141:231–237
Badaracco A, Garaicoechea L, Rodríguez D, Uriarte EL, Odeón A, Bilbao G, Galarza R, Abdala A, Fernandez F, Parreño V (2012) Bovine rotavirus strains circulating in beef and dairy herds in Argentina from 2004 to 2010. Vet Microbiol 158:394–399
Badaracco A, Garaicoechea L, Matthijnssens J, Louge Uriarte E, Odeon A, Bilbao G, Fernandez F, Parra GI, Parreno V (2013) Phylogenetic analyses of typical bovine rotavirus genotypes G6, G10, P[5] and P[11] circulating in Argentinean beef and dairy herds. Infect Genet Evol 18:18–30
Badiei K, Pourjafar M, Ghane M (2010) Fecal rotavirus antigen in diarrheic calves of. high and average producing holstein dairy cows. Global Vet 5:175–179
Barreiros MA, Alfieri AF, Medici KC, Leite JP, Alfieri AA (2004) G and P genotypes of group A rotavirus from diarrhoeic calves born to cows vaccinated against the NCDV (P[1], G6) rotavirus strain. J Vet Med B Infect Dis Vet Public Health 51:104–109
http://www.bioveta.eu/en/products/veterinary-products/kolibin-rc-neo-injection-suspension-for-cattle.html. Accessed 10 July 2016
Cashman O, Lennon G, Sleator RD, Power E, Fanning S, O’Shea H (2010) Changing profile of the bovine rotavirus G6 population in the south of Ireland from 2002 to 2009. Vet Microbiol 146:238–244
Chang KO, Parwani AV, Saif LJ (1996) The characterization of VP7 (G type) and VP4 (P type) genes of bovine group A rotaviruses from field samples using RT-PCR and RFLP analysis. Arch Virol 141:1727–1739
Das M, Dunn SJ, Woode GN, Greenberg HB, Rao CD (1993) Both surface proteins (VP4 and VP7) of an asymptomatic neonatal rotavirus strain (I321) have high levels of sequence identity with the homologous proteins of a serotype 10 bovine rotavirus. Virology 194:374–379
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469
Dhama K, Chauhan RS, Mahendran M, Malik SV (2009) Rotavirus diarrhea in bovines and other domestic animals. Vet Res Commun 33:1–23
Doan YH, Nakagomi T, Aboudy Y, Silberstein I, Behar-Novat E, Nakagomi O, Shulman LM (2013) Identification by full-genome analysis of a bovine rotavirus transmitted directly to and causing diarrhea in a human child. J Clin Microbiol 51:182–189
Eesteghamati A, Gouya M, Keshtkar A, Najafi L, Zali MR, Sanaei M, Yaghini F, El Mohamady H, Patel M, Klena JD, Teleb N (2009) Sentinel hospital-based surveillance of rotavirus diarrhea in Iran. J Infect Dis 200(Suppl 1):S244–S247
Estes MK, Kapikian AZ (2007) Fields virology, 5th edn. Lippincott Williams & Wilkins, Philadelphia
Falcone E, Tarantino M, Di Trani L, Cordioli P, Lavazza A, Tollis M (1999) Determination of bovine rotavirus G and P serotypes in Italy by PCR. J Clin Microbiol 37:3879–3882
Fodha I, Boumaiza A, Chouikha A, Dewar J, Armah G, Geyer A, Trabelsi A, Steele AD (2005) Detection of group a rotavirus strains circulating in calves in Tunisia. J Vet Med B Infect Dis Vet Public Health 52:49–50
Fukai K, Sakai T, Hirose M, Itou T (1999) Prevalence of calf diarrhea caused by bovine group A rotavirus carrying G serotype 8 specificity. Vet Microbiol 66:301–311
Fukai K, Maeda Y, Fujimoto K, Itou T, Sakai T (2002) Changes in the prevalence of rotavirus G and P types in diarrheic calves from the Kagoshima prefecture in Japan. Vet Microbiol 86:343–349
Garaicoechea L, Bok K, Jones LR, Combessies G, Odeon A, Fernandez F, Parreno V (2006) Molecular characterization of bovine rotavirus circulating in beef and dairy herds in Argentina during a 10-year period (1994–2003). Vet Microbiol 118:1–11
Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J, Das BK, Bhan MK (1992) Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol 30:1365–1373
Ghorbanpour M, Keyvanfar H, Shapouri S (2004) The dsRNA electrophoretype of some isolated Iranian. Arch Razi 58:85–89
Gomara MI, Cubitt D, Desselberger U, Gray J (2001) Amino acid substitution within the VP7 protein of G2 rotavirus strains associated with failure to serotype. J Clin Microbiol 39(10):3796–3798
Gouvea V, Santos N, Timenetsky Mdo C (1994) VP4 typing of bovine and porcine group A rotaviruses by PCR. J Clin Microbiol 32:1333–1337
Gouvea V, Santos N, Timenetsky Mdo C (1994) Identification of bovine and porcine rotavirus G types by PCR. J Clin Microbiol 32:1338–1340
Gunson RN, Miller J, Leonard A, Carman WF (2003) Importance of PCR in the diagnosis and understanding of rotavirus illness in the community. Commun Dis Public Health 6:63–65
Hassine-Zaafrane M, Sdiri-Loulizi K, Ben Salem I, Kaplon J, Ayouni S, Ambert-Balay K, Sakly N, Pothier P, Aouni M (2012) The molecular epidemiology of circulating rotaviruses: three-year surveillance in the region of Monastir, Tunisia. BMC Infect Dis 11:266
Hassine-Zaafrane M, Ben Salem I, Sdiri-Loulizi K, Kaplon J, Bouslama L, Aouni Z, Sakly N, Pothier P, Aouni M, Ambert-Balay K (2014) Distribution of G (VP7) and P (VP4) genotypes of group A bovine rotaviruses from Tunisian calves with diarrhoea. J Appl Microbiol 116:1387–1395
Holland RE (1990) Some infectious causes of diarrhea in young farm animals. Clin Microbiol Rev 3:345–375
Howe L, Sugiarto H, Squires RA (2008) Use of polymerase chain reaction for the differentiation of Group A bovine rotavirus G6, G8, and G10 genotypes in the North Island of New Zealand. N Z Vet J 56:218–221
Ishizaki H, Sakai T, Shirahata T, Taniguchi K, Urasawa T, Urasawa S, Goto H (1996) The distribution of G and P types within 117 isolates of bovine rotavirus in Japan. Vet Microbiol 48:367–372
Iturriza Gomara M, Wong C, Blome S, Desselberger U, Gray J (2002) Molecular characterization of VP6 genes of human rotavirus isolates: correlation of genogroups with subgroups and evidence of independent segregation. J Virol 76:6596–6601
Jere KC, Esona MD, Ali YH, Peenze I, Roy S, Bowen MD, Saeed IK, Khalafalla AI, Nyaga MM, Mphahlele J, Steele D, Seheri ML (2014) Novel NSP1 genotype characterised in an African camel G8P[11] rotavirus strain. Infect Genet Evol 21:58–66
Kaplon J, Fremy C, Bernard S, Rehby L, Aho S, Pothier P, Ambert-Balay K (2013) Impact of rotavirus vaccine on rotavirus genotypes and caliciviruses circulating in French cattle. Vaccine 31:2433–2440
Keyvanfar H, Ghorbanpoor M, Seify-abad Shapouri M (2001) A survey on prevalence of rotaviral diarrhea in dairy calves in Tehran region and determination of serotypes. J Faculty of Vet Med University of Tehran 3:3–11 (Persian)
Khalili B, Cuevas LE, Reisi N, Dove W, Cunliffe NA, Hart CA (2004) Epidemiology of rotavirus diarrhoea in Iranian children. J Med Virol 73:309–312
Lucchelli A, Kang SY, Jayasekera MK, Parwani AV, Zeman DH, Saif LJ (1994) A survey of G6 and G10 serotypes of group A bovine rotaviruses from diarrheic beef and dairy calves using monoclonal antibodies in ELISA. J Vet Diagn Invest 6:175–181
Madadgar O, Nazaktabar A, Keivanfar H, Zahraei Salehi T, Lotfollah Zadeh S (2015) Genotyping and determining the distribution of prevalent G and P types of group A bovine rotaviruses between 2010 and 2012 in Iran. Vet Microbiol 179:190–196
Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Palombo EA, Iturriza-Gomara M, Maes P, Patton JT, Rahman M, Van Ranst M (2008) Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J Virol 82:3204–3219
Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Banyai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gomara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreno V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M (2011) Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol 156:1397–1413
Matthijnssens J, Desselberger U (2012) Genome Diversity and Evolution of Rotaviruses. In: Hacker J, Dobrindt U, Kurth R (eds) Genome plasticity and infectious diseases. ASM Press, Washington, DC, pp 214–241
Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R (2012) VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol 157:1177–1182
Mayameii A, Shapouri MR, Ghorbanpour M, Hajikolaei MR, Keyvanfar H (2007) Molecular G typing of bovine rotaviruses in Iran. Pak J Biol Sci 10:3466–3469
Midgley SE, Banyai K, Buesa J, Halaihel N, Hjulsager CK, Jakab F, Kaplon J, Larsen LE, Monini M, Poljsak-Prijatelj M, Pothier P, Ruggeri FM, Steyer A, Koopmans M, Bottiger B (2012) Diversity and zoonotic potential of rotaviruses in swine and cattle across Europe. Vet Microbiol 156:238–245
Modarres S, Rahbarimanesh AA, Karimi M, Motamedi-Rad M, Sohrabi A, Nasiri-Oskoii N (2008) Electrophoretic RNA genomic profiles of rotavirus strains prevailing among hospitalized children with acute gastroenteritis in tehran, iran. Arch Iran Med 11:526–531
Monini M, Cappuccini F, Battista P, Falcone E, Lavazza A, Ruggeri FM (2008) Molecular characterization of bovine rotavirus strains circulating in northern Italy, 2003–2005. Vet Microbiol 129:384–389
Morshedi A (2004) Detection of Rotavirus infection in young calves by ELISA and comparison with complement fixation test. Ind Vet J 81:1189–1191
Naficy AB, Abu-Elyazeed R, Holmes JL, Rao MR, Savarino SJ, Kim Y, Wierzba TF, Peruski L, Lee YJ, Gentsch JR, Glass RI, Clemens JD (1999) Epidemiology of rotavirus diarrhea in Egyptian children and implications for disease control. Am J Epidemiol 150:770–777
Nakagomi O, Isegawa Y, Ward RL, Knowlton DR, Kaga E, Nakagomi T, Ueda S (1994) Naturally occurring dual infection with human and bovine rotaviruses as suggested by the recovery of G1P8 and G1P5 rotaviruses from a single patient. Arch Virol 137:381–388
Papp H, Laszlo B, Jakab F, Ganesh B, De Grazia S, Matthijnssens J, Ciarlet M, Martella V, Banyai K (2013) Review of group A rotavirus strains reported in swine and cattle. Vet Microbiol 165:190–199
Park SH, Saif LJ, Jeong C, Lim GK, Park SI, Kim HH, Park SJ, Kim YJ, Jeong JH, Kang MI, Cho KO (2006) Molecular characterization of novel G5 bovine rotavirus strains. J Clin Microbiol 44:4101–4112
Ramig RF (1997) Genetics of the rotaviruses. Annu Rev Microbiol 51:225–255
Reidy N, Lennon G, Fanning S, Power E, O’Shea H (2006) Molecular characterisation and analysis of bovine rotavirus strains circulating in Ireland 2002-2004. Vet Microbiol 117:242–247
Sdiri-Loulizi K, Gharbi-Khelifi H, de Rougemont A, Chouchane S, Sakly N, Ambert-Balay K, Hassine M, Guediche MN, Aouni M, Pothier P (2008) Acute infantile gastroenteritis associated with human enteric viruses in Tunisia. J Clin Microbiol 46:1349–1355
Simmonds MK, Armah G, Asmah R, Banerjee I, Damanka S, Esona M, Gentsch JR, Gray JJ, Kirkwood C, Page N, Iturriza-Gomara M (2008) New oligonucleotide primers for P-typing of rotavirus strains: Strategies for typing previously untypeable strains. J Clin Virol 42:368–373
Suzuki Y, Sanekata T, Sato M, Tajima K, Matsuda Y, Nakagomi O (1993) Relative frequencies of G (VP7) and P (VP4) serotypes determined by polymerase chain reaction assays among Japanese bovine rotaviruses isolated in cell culture. J Clin Microbiol 31:3046–3049
Swiatek DL, Palombo EA, Lee A, Coventry MJ, Britz ML, Kirkwood CD (2010) Detection and analysis of bovine rotavirus strains circulating in Australian calves during 2004 and 2005. Vet Microbiol 140:56–62
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Taniguchi K, Urasawa T, Pongsuwanna Y, Choonthanom M, Jayavasu C, Urasawa S (1991) Molecular and antigenic analyses of serotypes 8 and 10 of bovine rotaviruses in Thailand. J Gen Virol 72(Pt 12):2929–2937
Taniguchi K, Wakasugi F, Pongsuwanna Y, Urasawa T, Ukae S, Chiba S, Urasawa S (1992) Identification of human and bovine rotavirus serotypes by polymerase chain reaction. Epidemiol Infect 109:303–312
Theil KW, McCloskey CM (1995) Rotavirus shedding in feces of gnotobiotic calves orally inoculated with a commercial rotavirus-coronavirus vaccine. J Vet Diagn Invest 7:427–432
van der Heide R, Koopmans MP, Shekary N, Houwers DJ, van Duynhoven YT, van der Poel WH (2005) Molecular characterizations of human and animal group a rotaviruses in the Netherlands. J Clin Microbiol 43:669–675
Varshney B, Jagannath MR, Vethanayagam RR, Kodhandharaman S, Jagannath HV, Gowda K, Singh DK, Rao CD (2002) Prevalence of, and antigenic variation in, serotype G10 rotaviruses and detection of serotype G3 strains in diarrheic calves: implications for the origin of G10P11 or P11 type reassortant asymptomatic strains in newborn children in India. Arch Virol 147:143–165
Ward P, Poitras E, Leblanc D, Gagnon CA, Brassard J, Houde A (2013) Comparison of different RT-qPCR assays for the detection of human and bovine group A rotaviruses and characterization by sequences analysis of genes encoding VP4 and VP7 capsid proteins. J Appl Microbiol 114:1435–1448
Wierzba TF, Abdel-Messih IA, Abu-Elyazeed R, Putnam SD, Kamal KA, Rozmajzl P, Ahmed SF, Fatah A, Zabedy K, Shaheen HI, Sanders J, Frenck R (2006) Clinic-based surveillance for bacterial- and rotavirus-associated diarrhea in Egyptian children. Am J Trop Med Hyg 74:148–153
Wilde J, Yolken R, Willoughby R, Eiden J (1991) Improved detection of rotavirus shedding by polymerase chain reaction. Lancet 337:323–326
Zarnani AH, Modarres S, Jadali F, Sabahi F, Moazzeni SM, Vazirian F (2004) Role of rotaviruses in children with acute diarrhea in Tehran. Iran. J Clin Virol 29:189–193
Acknowledgments
This work was financially supported by research grant no. 2-18-18-88050 of Razi Vaccine and Serum Research Institute, Iran. A part of this research was performed at the National Reference Centre for Enteric Viruses, Dijon, France. Furthermore, this study was kindly supported by Center for International Scientific Studies and Collaboration (CISSC) and the French Embassy in Tehran. Lastly, we thank Amina Allalou and Dr. Naheed Mojgani for their contributions to English editing of this paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Pourasgari, F., Kaplon, J., Karimi-Naghlani, S. et al. The molecular epidemiology of bovine rotaviruses circulating in Iran: a two-year study. Arch Virol 161, 3483–3494 (2016). https://doi.org/10.1007/s00705-016-3051-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3051-0