
Abstract
A previously unidentified chicken parvovirus (ChPV) strain, associated with runting-stunting syndrome (RSS), is now endemic among chickens in China. To explore the genetic diversity of ChPV strains, we determined the first complete genome sequence of a novel ChPV isolate (GX-CH-PV-7) identified in chickens in Guang Xi, China, and showed moderate genome sequence similarity to reference strains. Analysis showed that the viral genome sequence is 86.4 %–93.9 % identical to those of other ChPVs. Genetic and phylogenetic analyses showed that this newly emergent GX-CH-PV-7 is closely related to Gallus gallus enteric parvovirus isolate ChPV 798 from the USA, indicating that they may share a common ancestor. The complete DNA sequence is 4612 bp long with an A+T content of 56.66 %. We determined the first complete genome sequence of a previously unidentified ChPV strain to elucidate its origin and evolutionary status.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Parvoviruses (family Parvoviridae) that infect vertebrate hosts make up the subfamily Parvovirinae, while those that infect arthropods make up the subfamily Densovirinae. The traditional classification of parvoviruses was recently replaced by eight genera (Amdoparvovirus, Aveparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Protoparvovirus, and Tetraparvovirus) by the International Committee for Taxonomy of Viruses (http://www.ictvonline.org/virusTaxonomy.asp). Chicken parvovirus (ChPV) and turkey parvovirus (TuPV) are classified as possible members of a different taxon, the new genus Aveparvovirus.
ChPV is a non-enveloped, positive-sense, single-stranded DNA virus [1] with a genome of approximately 5 kb containing three open reading frames (ORFs). ORF1 and ORF2 encode a non-structural (NS) protein, which is involved in viral replication, and the capsid proteins (VP1, VP2), respectively, while the function of ORF3 (NP1), located between ORF1 and ORF2, is unclear [2]. ChPV is highly prevalent in poultry flocks affected by runting-stunting syndrome (RSS), but it is also found in apparently healthy flocks [3]. RSS is characterized by stunted growth, diarrhea, immune dysfunction and increased mortality [4]. Economic losses arise from increased production costs due to poor feed conversion and the cost of treatment. The first ChPV sequence was reported by Day and Zsak in 2010 [2]. ChPV infection in chickens is endemic to the United States (US), Poland, Hungary, Croatia and South Korea [5–9]. However, only 10 complete genomic sequences have been submitted to GenBank so far: one from Hungary, four from the US and four from South Korea, [2, 10, 11]. In China, the prevalence of ChPV in chicken flocks is increasing, although the causative strain remains unidentified.
In the present study, we investigated the presence of ChPV strains in Guangxi province to establish a genetic database for ChPV genome sequences from chickens and to analyze the evolution of ChPVs. By determining the complete genome sequence of a previously unidentified ChPV strain, we elucidate its origin and evolutionary status and lay the foundation for the development of a new vaccine.
Viral DNA was extracted from tracheal and cloacal swabs (collected in 2014-2016 from 23 commercial farms in Guangxi, China) using EasyPure viral DNA/RNA kits (Transgen, Beijing, China), according to the manufacturer’s instructions. The non-structural (NS) gene was amplified by PCR, using primers (Supplementary Table 1) designed to target conserved regions (NS, 561 bp) first [9]. The positive samples were confirmed by partial sequencing of the NS1 gene. BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis of these samples revealed 98 %–100 % nt sequence identity to the ChPV ABU-P1 strain (genus Aveparvovirus, family Parvoviridae) isolated in Hungary. Based on an alignment of the complete sequences of three ChPV/TuPV isolates deposited in GenBank, three specific primers pairs were then designed to amplify the complete genome of one positive ChPV sample (GX-CH-PV-7) (Supplementary Table 1).
PCR amplifications were performed in 50 μL (total volume) containing 10 μL of 5× PrimeSTAR GXL Buffer (Mg2+ Plus), 0.5 μL of PrimeSTAR GXL DNA Polymerase, 4 μL of dNTP mixture (2.5 mM each), 0.5 μL of each primer (20 μM), 1 μL of DNA template, and 33.5 μL of RNase-free water. PCR products were separated by electrophoresis on 1.2 % agarose gels stained with gel red and visualized using an ultraviolet camera. The products were purified using a TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0 (TaKaRa). The purified PCR product was cloned into pMD-19T (TaKaRa) vector. Sanger sequencing was performed on an ABI 3730 DNA Analyzer (Takara Biotech Co., Beijing, China).
The complete genome of GX-CH-PV-7 (GenBank accession no. KU523900) contains 4,612 bp with three ORFs. ORF1 encodes a nonfunctional protein NS1 (nt 1-2085, 695 aa), ORF3 (nt 2,283–2,587, 101 aa) encodes a putative protein NP1, and ORF2 encodes the minor and major capsid proteins, VP1 (nt 2,585-4612, 676 aa) and VP2 (nt 3,002–4,612, 537aa), respectively. BLAST analysis of the whole GX-CH-PV-7 genome revealed 92 % nt sequence identity to ChPV ABU-P1, thus confirming the identity of the isolate as a chicken parvovirus.
Analysis of complete and partial genomic sequences using ClustalW (http://www.clustal.org) and MEGA 6.0.6 [12] software indicated that GX-CH-PV-7 shares the highest identity (93.9 %) with ChPV 367 (USA) and 89.0 %–93.8 % identity with other ChPV strains isolated in the USA, Hungary and South Korea. Moreover, compared with the regional sequences of 10 other ChPV strains, NS1 of ChPV shared 93.1 %–96.1 % nt and 87.8 %–98.7 % aa sequence identity; NP1 shared 72.0 %–90.4 % nt and 90.2 %–95.1 % aa sequence identity; VP1 shared 88.9 %–92.4 % nt and 94.8 %–96.8 % aa sequence identity, and VP2 shared 83.4 %–91.4 % nt and 93.5 %–97.0 % aa sequence identity.
A well-conserved phosphate-binding loop (P-loop) motif (NS1 of ABU-P1 at 392–399 aa) and a NTP-binding motif (NS1 of ABU-P1 at 436-437 aa) were present in all isolates and the ABU-P1 strain [2]. A Z-test of selection revealed that the NS1 genes of GX-CH-PV-7 and ChPV ABU-P1 had undergone purifying selection. Putative nt and aa motifs, including the start codons for VP1 (nt 2,585–2,587) and VP2 (nt 3,002–3,004), the glycine-rich sequence (aa 164–172; VP1), the VP2 leucine residue (aa 291; VP1), and the fivefold cylinder region (aa 273–290; VP1) were identified, which were identified based on multiple alignments of ChPVs (Fig. 1a). A third start codon with a favorable translation initiation context (GACATGG) was found at position 3,194 in the GX-CH-PV-7 genome, as well as in ChPV ABU-P1, ChPV 736, ChPV 798, and ChPV 367, and this may represent the beginning of a putative VP3 ORF, as has been described in goose and Muscovy duck parvoviruses [13] (Fig. 1b).

Structural motifs in ORF2 and Simplot analysis of ChPVs. a Putative VP3 start codons among 11 ChPVs. b VP2 amino acid sequences comprising the fivefold cylinders in VP2 proteins. Shaded letters indicate the structural motif composed of fivefold cylinder regions. The conserved leucine (shown in a box) represents the structural motif for the constriction of the pore in cylindrical projections at each fivefold axis of symmetry. Both the nucleotide and amino acid positions are from the ABU-P1 strain [2]. c Simplot analysis based on full-length nucleotide sequences using GX-CH-PV-7 as the query sequence
Using SimPlot 3.5.1 (http://sray.med.som.jhmi.edu/SCRoftware/simplot), we observed moderate nucleotide sequence similarity between GX-CH-PV-7 and the other 10 ChPV strains isolated in the USA (ChPV 367, ChPV 798, ChPV 736, ChPV 841), Hungary (ChPV ABU P1) and South Korea (ChPV ADL120686, ChPV ADL120019, ChPV ADL120035; ChPV ParvoD62/2013, ChPV ParvoD11/2007) at the whole-genome level, except at the 3’ end of ORF2 (encoding VP1 and VP2), which showed less similarity among ChPV/TuPV strains. Genomic similarity was highest at the 5’ end of the genome, except in the VP1 region of South Korea ChPV ParvoD11/2007 (South Korea) and ChPV 841 (USA). We found no evidence of recombination between GX-CH-PV-7 and the other 10 ChPV strains [14], indicating that GX-CH-PV-7 was not generated through recombination from ChPVs (Fig. 1c).
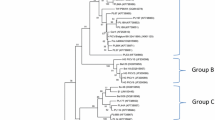
Neighbor-joining trees were constructed with the substitution model of Tamura and Nei in MEGA 6.0.6; 1,000 bootstrap replicates were used to test the reliability of the tree topology. Phylogenetic analysis of the complete ChPV genome sequence revealed three major clusters. The GX-CH-PV-7 strain segregated into a distinct branch separate from other ChPVs and appeared to have a close relationship to Gallus gallus enteric parvovirus isolate ChPV 798 strain from the USA (GenBank accession nos. KM598416), forming a second cluster (Fig. 2).

Phylogenetic neighbor-joining tree based on near-complete genomic sequences of GX-CH-PV and 17 other ChPV/TuPV isolates. GenBank accession numbers follow the names of ChPV/TuPV strains. The numbers near the branches indicate the confidence level calculated by bootstrapping (n = 1,000). The length of each pair of branches represents the distance between sequence pairs. Scale bar represents 0.01 nt substitutions per site. “◆” and “■” represent ChPV and TuPV strains, respectively, isolated in China (GX-Tu-PV-2 data not shown)
Although ChPV strain GX-CH-PV-7 is closely related to Gallus gallus enteric parvovirus isolate ChPV 798 (genus, Aveparvovirus) from the USA, it is not known, when and how this virus was introduced into China. Further investigations are required to determine the impact of GX-CH-PV-7 on RSS in commercial chickens. Moreover, whole-genome sequence analysis should be performed for other strains from different locations to determine whether the virus was introduced into China by a single entry or multiple entries. Cases of parvovirus infection in chickens have not been reported in China. No apparent full-scale outbreaks of acute or chronic RSS have occurred. Studies on the geographic prevalence and genetic diversity of ChPV and cross-species infection are lacking, and studies on the zoonotic properties of ChPV are incomplete but underway. Knowledge of the diffusion pattern of ChPV around China is also lacking. Given these facts, ChPV surveillance is essential in China.
In conclusion, this study provides the first complete nucleotide sequence of ChPV strain GX-CH-PV-7 from a chicken in China. Genetic and phylogenetic analyses demonstrate that GX-CH-PV-7 is closely related to a ChPV isolate from the USA, suggesting that they may share a common parentage. Given the economic loss caused by ChPV infection in commercial chickens, an extensive epidemiological study of the geographic distribution of ChPV in commercial chicken flocks in China is needed. The data of this study can be referenced to design ChPV diagnostic assays and facilitate further ChPV epidemiological studies in China.
References
Kisary J, Avalosse B, Miller-Faures A, Rommelaere J (1985) The genome structure of a new chicken virus identifies it as a parvovirus. J Gen Virol 66(Pt 10):2259–2263
Day JM, Zsak L (2010) Determination and analysis of the full-length chicken parvovirus genome. Virology 399:59–64
Pass DA, Robertson MD, Wilcox GE (1982) Runting syndrome in broiler chickens in Australia. Vet Rec 110:386–387
Guy JS (1998) Virus infections of the gastrointestinal tract of poultry. Poult Sci 77:1166–1175
Bidin M, Lojkic I, Bidin Z, Tiljar M, Majnaric D (2011) Identification and phylogenetic diversity of parvovirus circulating in commercial chicken and turkey flocks in Croatia. Avian Dis 55:693–696
Domanska-Blicharz K, Jacukowicz A, Lisowska A, Minta Z (2012) Genetic characterization of parvoviruses circulating in turkey and chicken flocks in Poland. Arch Virol 157:2425–2430
Koo BS, Lee HR, Jeon EO, Han MS, Min KC, Lee SB, Mo IP (2013) Molecular survey of enteric viruses in commercial chicken farms in Korea with a history of enteritis. Poult Sci 92:2876–2885
Palade EA, Kisary J, Benyeda Z, Mandoki M, Balka G, Jakab C, Vegh B, Demeter Z, Rusvai M (2011) Naturally occurring parvoviral infection in Hungarian broiler flocks. Avian Pathol: J WVPA 40:191–197
Zsak L, Strother KO, Day JM (2009) Development of a polymerase chain reaction procedure for detection of chicken and turkey parvoviruses. Avian Dis 53:83–88
Koo BS, Lee HR, Jeon EO, Han MS, Min KC, Lee SB, Bae YJ, Cho SH, Mo JS, Kwon HM, Sung HW, Kim JN, Mo IP (2015) Genetic characterization of three novel chicken parvovirus strains based on analysis of their coding sequences. Avian Pathol: J WVPA 44:28–34
Zsak L, Cha RM, Li F, Day JM (2015) Host specificity and phylogenetic relationships of chicken and turkey parvoviruses. Avian Dis 59:157–161
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Zadori Z, Stefancsik R, Rauch T, Kisary J (1995) Analysis of the complete nucleotide sequences of goose and muscovy duck parvoviruses indicates common ancestral origin with adeno-associated virus 2. Virology 212:562–573
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Acknowledgments
We acknowledge and appreciate the excellent technical help provided by several staff in cities local to the Animal Disease Prevention and Control Center, Guangxi, for assistance with sample collection. This study was funded by the Guangxi Science and Technology Bureau (1222003-2-4) and the Guangxi Government Senior Scientist Foundation (2011B020) (Guangxi, China).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that we have no conflict of interest.
Ethical approval
This study was approved by the Institutional Animal Care and Use Committee (IACUC) of the Guangxi Veterinary Research Institute. Briefly, all the chickens were given ad libitum access to feed and water. Tracheal and cloacal swab samples were gently collected from the live chickens as per IACUC protocol to minimize animal suffering.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Feng, B., Xie, Z., Deng, X. et al. Genetic and phylogenetic analysis of a novel parvovirus isolated from chickens in Guangxi, China. Arch Virol 161, 3285–3289 (2016). https://doi.org/10.1007/s00705-016-2999-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2999-0