
Abstract
Begomoviruses are whitefly-transmitted, single-stranded DNA viruses that infect a variety of cultivated (crop) and non-cultivated (weed) plants. The present study identified a novel begomovirus and satellites (alpha- and betasatellite) in Senna occidentalis (syn. Cassia occidentalis) showing leaf curl symptoms. The begomovirus shared a maximum sequence identity of 88.6 % with french bean leaf curl virus (JQ866297), whereas the alphasatellite and the betasatellite shared identities of 98 % and 90 % with ageratum yellow vein India alphasatellite (LK054802) and papaya leaf curl betasatellite (HM143906), respectively. No other begomovirus or satellites were detected in the suspected plants. We propose to name the virus “senna leaf curl virus” (SenLCuV).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The family Geminiviridae is divided into seven genera – Becurtovirus, Begomovirus, Curtovirus, Eragrovirus, Mastrevirus, Topocuvirus and Turncurtovirus – based on phylogenetic relationship, genome organization, insect vector and host range [1]. Begomoviruses constitute the largest genus in the family Geminiviridae and are transmitted by whiteflies of the species Bemisia tabaci to a wide range of cultivated and noncultivated dicot plants [2]. Several begomoviruses are known to infect legume crops and causes significant yield loss [3]. Some of the begomoviruses have also been reported from leguminous weeds such as Senna occidentalis, Leucaena leucocephala and Rhynchosia minima [4, 5]. The weeds serve as alternative hosts and function as hotspots of recombination, leading to the evolution of new viruses or strains [4]. In the present study, we identified a novel begomovirus and associated satellites (alpha- and betasatellites), in Senna occidentalis (syn. Cassia occidentalis).
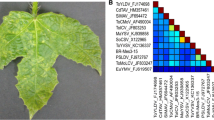
Leaf samples from S. occidentalis plants showing curling of leaves (Fig. 1B) were collected in the vicinity of agricultural fields in Mohali, India (Latitude: 30° 42′ 45.1″ North; Longitude: 76° 42′ 30.7″ East). Leaf sample from a total of 20 symptomatic and five asymptomatic plants were collected and stored at -80 °C. Based on the symptoms, begomovirus infection was suspected. Total DNA was isolated from leaf tissue using the CTAB method [6]. Rolling-circle amplification (RCA) was performed – using total DNA as template and Φ29 polymerase – to amplify viral DNA (Illustra TempliPhi Kit, GE Healthcare). RCA products were digested by BamHI, HindIII, KpnI, NdeI, PstI, SacI and XbaI restriction endonucleases (New England Biolabs, UK) and loaded onto an agarose gel. Gel electrophoresis of RCA products digested with BamHI, HindIII, KpnI and XbaI yielded the expected fragments of ~2.7 kb, corresponding to the genome size of a begomovirus (Fig. 1C), whereas the digestion with NdeI, PstI and SacI yielded fragments of ~1.3 kb, corresponding to the size of a satellite (Fig. 1D). The asymptomatic plants showed the absence of any band representing a viral or satellite DNA (Fig. 1C and D). A total of 18 out of 20 symptomatic plant samples yielded fragments corresponding to the size of a begomovirus (~2.7 kb) and the satellites (~1.3 kb). The RCA fragments (~2.7 and 1.3 kb) obtained using the restriction enzymes (BamHI, HindIII, KpnI, NdeI, PstI, SacI and XbaI) were cloned separately in the SK+ cloning vector (Agilent Technologies), yielding seven types of clones for each positive sample. Two clones for each restriction enzyme fragment (2 × 7 = 14) were sequenced partially, using M13 universal primers, from each of the 18 positive samples. Upon partial sequencing, a total of 18 plants tested positive for the virus and the betasatellite, and a total of 15 plants tested positive for the virus, the alphasatellite and the betasatellite. Partial sequencing of the 252 (14 × 18 = 252) clones confirmed the presence of a single type of begomovirus, alphasatellite and betasatellite. Five random clones each for the virus, alphasatellite and betasatellite were sequenced using primer walking to determine the full-length genomic sequence.

Asymptomatic (A) and symptomatic (B) S. occidentalis plants. Symptomatic plants showing downward curling of leaves (B). Restriction digestion of the rolling-circle-amplified (RCA) DNA (C, D). Lanes 1 and 2 of panel C show RCA products digested by KpnI, and the lanes 1 and 2 of panel D show RCA DNA digested by SacI. Lane M in panels C and D shows the size markers (λ DNA digested by EcoRI/HindIII)
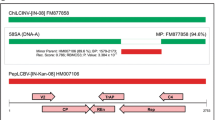
Sequence analysis of the viral clones from five different samples, using sequence demarcation tool (SDT) [7], revealed the presence of highly similar sequences (Supplementary Table 1). The complete nucleotide sequence of a viral clone (SenLCuV:Moh6:2013) was submitted to the GenBank database (KU852742). The full-length viral genome was 2742 bp long and displayed a typical monopartite begomovirus genome organization. The viral genome had two open reading frames (ORFs), V1 and V2, on the viral-sense strand and four ORFs (C1, C2, C3 and C4) on the complementary-sense strand. In addition, it contained a probable ORF, C5 (Supplementary Figure 1A). The conserved nonanucleotide sequence (TAATATTAC) was present within the region reported to be the origin of replication (Supplementary Figures 1A and B). Comparison of the predicted iterons from other begomoviruses with the corresponding region in the virus under study suggested that CCGCT is the probable iteron (Supplementary Figure 1B). Pairwise identity analysis (Supplementary Table 2), using SDT [7], showed that the begomovirus under study shares the highest nucleotide sequence identity of 88.6 % with french bean leaf curl virus (JQ866297) and corchorus yellow vein mosaic virus (KC223600). This is below the threshold of 91 % [2] and thus qualifies this isolate to be considered a member of a new begomovirus species.
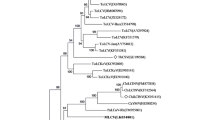
Phylogenetic analysis of the begomovirus under study was performed using the neighbor-joining method with 1000 bootstrap replicates in MEGA 6.0 [8]. The tree was rooted using an outlier sequence representing a different begomovirus species. Phylogenetic analysis revealed that the begomovirus under study formed a separate cluster and showed only a distant relationship to the previously known begomoviruses (Fig. 2). Thus, the begomovirus detected in this study should be considered a member of a new species. We propose to name this virus “senna leaf curl virus” (SenLCuV).

Dendrogram showing the relationship between SenLCuV (indicated by **) and other begomoviruses. The percentage of replicate trees in which the associated sequences clustered together in the bootstrap test (1000 replicates) is shown next to the branches
Begomoviruses are known to have a high recombination rate and evolve rapidly through genetic recombination [9]. To identify putative recombinant sites, recombination analysis was performed with aligned sequences of probable parents using MUSCLE in MEGA 6.0 [8]. The resultant alignment was exported as a FASTA file and used for predicting recombinant fragments in Recombination Detection Program version 4 [10]. The methods, RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan and 3Seq were used to scan the recombination events and identify probable parental sequences and recombination breakpoints. A recombination event detected by five methods with p-values less than 0.05 was considered for further analysis. Analysis suggested that the putative fragment was 1431 nucleotides long and covered a part of the C1 ORF and the whole V1, C3, C2 and C5 ORFs (Supplementary Figure 1C). The major and minor parents resembled pedilanthus leaf curl virus (PeLCV) and cotton leaf curl Kokhran virus (CLCuKV).
Sequencing of ~1.3-kb RCA restriction fragments resulted in identification of an alphasatellite and a betasatellite. All five clones gave highly similar sequences for the alphasatellite and betasatellite. The alphasatellite and betasatellite were also detected by polymerase chain reaction (PCR) amplification using the primer pairs nanofor/nanorev and β01/04, respectively [11]. The alphasatellite (KU852743) and betasatellite (KU852744) were 1389 and 1362 bp long, respectively. The alphasatellite showed 98 % sequence identity to ageratum yellow vein India alphasatellite (AYVIA; LK054802). AYVIA detected in this study contained all of the features of an alphasatellite, including the presence of a Rep gene and an A-rich region (Supplementary Figure 1A). The betasatellite showed 90 % sequence identity to papaya leaf curl betasatellite (PaLCuB; HM143906). The betasatellite contained all of the features of betasatellites, including the satellite-conserved region (SCR), the A-rich region and the βC1 gene (Supplementary Figure 1A). It also contained the conserved nonanucleotide sequence (TAATATTAC) that is common among the betasatellites and begomoviruses (Supplementary Figure 1A and C).
Although alphasatellites depend on helper-virus-encoded proteins for movement within and between plants, they are capable of autonomous replication in permissible plant cells, due to their own Rep proteins [12]. On the other hand, betasatellites are completely dependent on their helper begomoviruses. Betasatellites are predicted to contain the iteron-like sequences that are involved in trans-replication compatibility of betasatellites with different helper viruses [13, 14]. The iteron-like sequences present in betasatellites are suggested to functionally mimic the iterons of the helper begomoviruses [13, 14]. Upon analysis, we found the presence of iteron-like sequences in the PaLCuB genome (Supplementary Figure 1B). These may be required for trans-replication of PaLCuB by SenLCuV in the host plant.
Here, we report the presence of a novel begomovirus, SenLCuV, a betsatellite, PaLCuB, and an alphasatellite, AYVIA, in a legume weed, S. occidentalis. Recombination analysis indicated that SenLCuV might have evolved through recombination between viruses resembling PeLCV and CLCuKV. However, neither of these two viruses (PeLCV and CLCuKV) was detected in S. occidentalis during our study. A detailed study on a larger number of samples collected from across the country needs to be carried out to determine if the recombination took place in S. occidentalis or, alternatively, if it took place in another plant, followed by transmission of SenLCuV to S. occidentalis. S. occidentalis is prevalent across the country and grows perennially [15]. Hence, it may serve as a constant source of inoculum for whiteflies to spread the virus to other compatible hosts. The presence of SenLCuV in other crops remains to be surveyed to understand the epidemiology and host range of SenLCuV.
References
Adams MJ, King AMQ, Carstens EB (2013) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch Virol 158:2023–2030. doi:10.1007/s00705-013-1688-5
Brown JK, Zerbini FM, Navas-Castillo J et al (2015) Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160:1593–1619. doi:10.1007/s00705-015-2398-y
Hema M, Sreenivasulu P, Patil BL et al (2014) Tropical food legumes. Virus diseases of economic importance and their control. Adv Virus Res 90:431–505. doi:10.1016/B978-0-12-801246-8.00009-3
Alabi OJ, Ogbe FO, Bandyopadhyay R et al (2008) Alternate hosts of African cassava mosaic virus and East African cassava mosaic Cameroon virus in Nigeria. Arch Virol 153:1743–1747. doi:10.1007/s00705-008-0169-8
Ilyas M, Qazi J, Mansoor S, Briddon RW (2009) Molecular characterisation and infectivity of a “Legumovirus” (genus Begomovirus: family Geminiviridae) infecting the leguminous weed Rhynchosia minima in Pakistan. Virus Res 145:279–284. doi:10.1016/j.virusres.2009.07.018
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem bull 19:11–15
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9(9):e108277. doi:10.1371/journal.pone.0108277
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Rey MEC, Ndunguru J, Berrie LC et al (2012) Diversity of dicotyledenous-infecting geminiviruses and their associated DNA molecules in Southern Africa, including the South-west Indian Ocean Islands. Viruses 4:1753–1791. doi:10.3390/v4091753
Martin DP, Murrell B, Golden M et al (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1(1):vev003. doi:10.1093/ve/vev003
Kumar J, Kumar A, Roy JK et al (2010) Identification and molecular characterization of begomovirus and associated satellite DNA molecules infecting Cyamopsis tetragonoloba. Virus Genes 41:118–125. doi:10.1007/s11262-010-0482-7
Briddon RW, Stanley J (2006) Subviral agents associated with plant single-stranded DNA viruses. Virology 344:198–210. doi:10.1016/j.virol.2005.09.042
Nawaz-ul-Rehman MS, Mansoor S, Briddon RW, Fauquet CM (2009) Maintenance of an old world betasatellite by a new world helper begomovirus and possible rapid adaptation of the betasatellite. J Virol 83:9347–9355. doi:10.1128/JVI.00795-09
Kumar J, Kumar J, Singh SP, Tuli R (2014) Association of satellites with a mastrevirus in natural infection: complexity of Wheat dwarf India virus disease. J Virol 88:7093–7104. doi:10.1128/JVI.02911-13
Yadav JP, Arya V, Yadav S et al (2010) Cassia occidentalis L.: a review on its ethnobotany, phytochemical and pharmacological profile. Fitoterapia 81:223–230. doi:10.1016/j.fitote.2009.09.008
Acknowledgments
The authors are grateful to the Council of Scientific and Industrial Research and Department of Biotechnology, Government of India, for fellowships and funds, and to the Department of Science and Technology for JC Bose Fellowship to RT.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, J., Alok, A., Kumar, J. et al. Senna leaf curl virus: a novel begomovirus identified in Senna occidentalis . Arch Virol 161, 2609–2612 (2016). https://doi.org/10.1007/s00705-016-2931-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2931-7