
Abstract
Porcine circovirus type 2 (PCV2) is the etiological agent associated with several pig diseases that are collectively referred to as porcine circovirus-associated disease (PCVAD). Unfortunately, PCV2 has had a serious economic impact on the swine industry. In this study, we report the genome sequence of a novel PCV2 isolate (JS2015) identified in pigs in Jiang Su, China. The complete DNA sequence was 1766 nucleotides long with an A+T content of 52.7 %. It lacked a guanine (G) at nucleotide position 1045 compared to other reference PCV2 strains with a sequence length of 1766 nucleotides. Genetic characterization and phylogenetic analysis showed that the isolate JS2015 was most closely related to members of the PCV2d (AY181946) lineage. Our data provide insight into the epidemiology of porcine circovirus and may facilitate further study of the origin and evolution of PCV2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine circovirus (PCV) is a member of the genus Circovirus of the family Circoviridae [1, 16, 17]. Based on antigenic properties, pathogenicity, and nucleotide composition, PCV can be divided into two groups: non-pathogenic viruses in the PCV1 group, and members of the PCV2 group, which are widely recognized as the causative agents of postweaning multisystemic wasting syndrome (PMWS) [2, 3, 13]. PCV2, which causes PCVAD, has become a global swine malady with a severe economic impact on the swine industry worldwide.
PCV2 has a single-stranded circular DNA genome [4] that contains three major open reading frames (ORF1, 2, 3): ORF1 encodes the replication-associated proteins (Reps) [6], ORF2 encodes the viral capsid proteins (Cap) [7, 14], and ORF3 encodes an apoptotic protein [18]. The genome size of PCV2 isolates with sequences in the GenBank database is 1766, 1767, or 1768 nucleotides, and of the three, strains with 1766 nucleotides are the least frequent. So far, 15 strains with a sequence length of 1766 nucleotides have been identified worldwide, the first of which was reported by Dupont et al in Denmark in 2008 [9].
Three PCV2 genotypes (PCV2a, b, and c) were described before 2010 [8], but more recently, four PCV2 genotypes (PCV2a, b, c, and d) have been recognized based on phylogenetic analysis [5, 12]. The first PCV2d sequence was reported in 2010 by Guo et al. [19].
The main objective of this study was to survey the presence of PCV2 strains in Jiang Su province, establish a genetic database for PCV2 genome sequences from these animals, and analyze the evolution of PCV2. This study will contribute to controlling the spread of disease and laying the foundation for the development of a new vaccine.
Provenance of the virus material
Total viral DNA was extracted from serum samples (collected in 2014 and 2015 in Jiang Su, China) using a QIAamp Viral DNA Mint Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. PCR analysis was used to amplify and sequence the full-length PCV2 genome. The sequences of the specific primers were as follows: F1, 5′-TCCGTGGATTGTTCTGTAGCA-3′; R1, 5′-GGAAGGGGGCCAGTTCGTCAC-3′; F2, 5′-ACTACTCCTCCCGCCATAC-3′; R2, 5′-GGAGGAGTAGTTTACATAGGGG-3′ [5, 13, 15].
PCR amplifications were performed in 25 μl (total volume) containing 12.5 μl of 2× Taq Plus Master Mix, 0.5 μl of each primer, 2 μl of DNA template and 9.5 μl of RNase-free water. PCR reactions were carried out in a Mastercycler (Eppendorf).
PCR products were analyzed by electrophoresis on 1.5 % agarose gels stained with ethidium bromide (EB), and visualized using an ultraviolet camera. The products were purified using a QIAquick PCR Purification Kit (QIAGEN). The purified PCR product was cloned into pMD-19T (TaKaRa) vector.
Sanger sequencing was performed on an automated sequencer (ABI 3730 DNA Analyzer), and sequence analysis and alignment analysis were performed using DNAMAN software and the online BLAST program (http://www.ncbi.nlm.nih.gov/Blast.cgi). To avoid artifacts produced by amplification, the entire procedure from PCR to sequencing was repeated twice. The sequence from this study was compared to the available reference PCV2 sequences in GenBank that had a sequence length of 1766. MEGA version 5.0 was used to analyze nucleotide and amino acid sequence similarity and diversity and to assess phylogenetic relationships by the neighbor-joining (NJ) method. The obtained sequence was submitted to GenBank.
Sequence analysis
The JS2015 sequence determined in this study was 1766 nucleotides in length and had an AT content of 52.7 %. Multiple sequence alignment analysis showed that this new sequence lacked a guanine (G) at nucleotide position 1045 compared to the other 15 PCV2 strains with a genome length of 1766 nt. Homology-and-distance matrices showed that these 16 nucleotide sequences varied between 94.7 % (GenBank: HQ831519) and 99.8 % (GenBank: HM038030), whereas the predicted amino acid (aa) sequence identity values for the full genomes were 86.7 %-98.9 %.
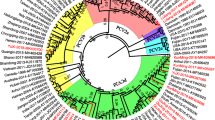
A phylogenetic tree was constructed that included the JS2015 sequence from this study and four prototype PCV2 strains from the GenBank database representing each of the PCV2 genotypes (accession numbers: AF055392, AF055394, EU148503, AY181947) (Fig. 1). In the phylogenetic tree, JS2015 grouped with isolates of the PCV2d genotype, supported by high confidence values.

Phylogenetic analysis of JS2015 compared with reference PCV2a, PCV2b, PCV2c, and PCV2d strains. An unrooted neighbor-joining (NJ) tree was constructed from aligned nucleotide sequences of the JS2015 strain and reference strains available in GenBank. The JS2015 strain identified in this study is indicated in bold inside a box
The genome of the JS2015 strain showed 95.3 %, 96.2 %, 94.3 %, and 98.1 % sequence identity to PCV2a, PCV2b, PCV2c, and PCV2d, respectively. JS2015 ORF1 (942 nt, encoding a protein of 313 aa) was 97.9 %, 97.1 %, 97.6 %, and 98.7 % identical, while JS2015 ORF2 (705 bp, encoding a protein of 234 aa) was 90.8 %, 94.7 %, 89.3 %, and 97.0 % identical to the corresponding ORF of PCV2a, PCV2b, PCV2c, and PCV2d, respectively. The predicted aa sequence of the protein encoded by ORF1 was 99.7 %, 99.5 %, 99.4 %, 99.6 % identical, and that of ORF2 was 97.6 %, 98.4 %, 96.8 %, 99.0 % identical, to those of PCV2a, b, c, and d, respectively. Further comparison of JS2015 ORF2 to the PCV2d reference strain showed that JS2015 lacked a G at position 1044, resulting in a shift of the stop codon compared to other PCV2d isolates, which normally encode a 233-aa protein (Fig. 2).

Characteristic nucleotide (nt) and deduced aa sequences of ORF2 of a PCV2d reference strain (GenBank: AY181946) and isolate JS2015. The JS2015 ORF2 lacks a guanine (G) at nucleotide position 1045, with the next A advanced to the position occupied by G in the PCV2d reference strain. This change leads to a shift of the stop codon from GUA to UAA at the end of ORF 2, resulting in one additional amino acid
In conclusion, genetic analysis of the nucleotide and deduced aa sequences of the JS2015 strain demonstrated that this newly identified strain was similar to other PCV2d strains previously reported in China. The complete genomic sequence of JS2015 was deposited in the GenBank database under accession no. KT220420. JS2015 has distinct genetic characteristics. However, several reports have demonstrated that infection with PCV2b/rBDH (HM038017), whose ORF2 encodes a 234-aa protein, results in more-severe clinical signs and lesions compared to infections with the main prevailing genotypes PCV2a and PCV2b [10, 11]. It is currently unknown whether this observation also extends to the JS2015 strain identified here. Therefore, further studies are needed to investigate the virulence of JS2015 mutant.
References
Tischer I, Rasch R, Tochtermann G (1974) Characterization of papovavirus and picornavirus-like particles in permanent pig kidney cell lines. Zentralbl Bakteriol Orig A 226(2):153–167
Hamel LA, Lin LL, Nayar GP (1998) Nucleotide sequence of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. J Virol 72(6):5262–5267
Ellis J, Hassard L, Clark E, Hardling J, Allan G, Wilson P, Strokappe J, Martin K, McNeilly F, Meehan B, Todd D, Haines D (1998) Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can Vet J 39(1):44–51
Tischer I, Gelderblom H, Vettermann W, Koch MA (1982) A very small porcine virus with circular single-stranded DNA. Nature 295(5844):64–66
Franzo G, Cortey M, de Castro AM, Piovezan U, Seqales J, Driqo M, Richtzenhain LJ (2015) Genetic characterisation of porcine circovirus type 2 (PCV2) strains from feral pigs in the Brazilian Pantanal: an opportunity to reconstruct the history of PCV2 evolution. Vet Microbiol 178(1–2):158–162
Mankertz A, Mankertz J, Wolf K, Buhk HJ (1998) Identification of a protein essential for replication of porcine circovirus. J Gen Virol 79(Pt2):381–384
Nawagitgul P, Morozov I, Bolin SR, Harms PA, Sorden SD, Paul PS (2000) Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol 81(Pt9):2281–2287
Segales J, Olvera J, Grau-Roma L, Charreyre C, Nauwynck H, Larsen L, Dupont K, McCullough K, Ellis J, Krakowka S, Mankertz A, Fredholm M, Fossum C, Timmusk S, Stockhofe Zurwieden N, Beattie V, Armstrong D, Grassland B, Baekbo P, Allan G (2008) PCV2 genotype definition and nomenclature. Vet Rec 162(26):867–868
Dupont K, Nielsen EO, Baekbo P, Larsen LE (2008) Genomic analysis of PCV2 isolates from Danish archives and a current PMWS case-control study supports a shift in genotypes with time. Vet Microbiol 128(1–2):56–64
Xiao CT, Halbur PG, Opriessnig T (2012) Complete genome sequence of a novel porcine circovirus type 2b variant present in cases of vaccine failures in the United States. J Virol 86(22):12469–12473
Guo L, Fu Y, Wang Y, Lu Y, Wei Y, Tang Q, Fan P, Liu J, Zhang L, Zhang F, Huang L, Liu D, Liu C (2012) A porcine circovirus type 2 (PCV2) mutant with 234 amino acids in capsid protein showed more virulence in vivo, compared with classical PCV2a/b strain. PLoS One 7(7):e41463. doi:10.1371/journal.pone.0041463
Xiao CT, Halbur PG, Opriessnig T (2015) Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J Gen Virol. doi:10.1099/vir.0.000100
Csagola A, Kecskemeti S, Kardos G, Kiss I, Tuboly T (2006) Genetic characterization of type 2 porcine circoviruses detected in Hungarian wild boars. Arch Virol 151(3):495–507
Seo HW, Park C, Kang I, Choi K, Jeong J, Park SJ, Chae C (2014) Genetic and antigenic characterization of a newly emerging porcine circovirus type 2b mutant first isolated in cases of vaccine failure in Korea. Arch Virol 159(11):3107–3111
An DJ, Roh IS, Song DS, Park CK, Park BK (2007) Phylogenetic characterization of porcine circovirus type 2 in PMWS and PDNS Korean pigs between 1999 and 2006. Virus Res 129(2):115–122
Cheung AK, Lager KM, Kohutyuk OI, Vincent AL, Henry SC, Baker RB, Rowland RR, Dunham AG (2007) Detection of two porcine circovirus type 2 genotypic groups in United States swine herds. Arch Virol 152(5):1035–1044
Pringle CR (1999) Virus taxonomy at the XIth International Congress of Virology, Sydney, Australia. Arch Virol 144(10):2065–2070
Wen LB, Wang FZ, He KW, Li B, Wang XM, Guo RL, Xie JP (2015) Recombination in truncated genome sequences of porcine circovirus type 2. Arch Virol 160(1):371–374
Guo J, Lu YH, Wei W, Huang LP, Liu M (2010) Porcine circovirus type 2 (PCV2): genetic variation and newly emerging genotypes in China. J Virol 7:273
Acknowledgments
This study was funded by the National Natural Science Foundation of China (31272574,30972184) and Fund for Independent Innovation of Agricultural Sciences in Jiangsu province CX(14)2045.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, D., He, K., Wen, L. et al. Genetic and phylogenetic analysis of a new porcine circovirus type 2 (PCV2) strain in China. Arch Virol 160, 3149–3151 (2015). https://doi.org/10.1007/s00705-015-2615-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2615-8