
Abstract
Development an effective vaccine may offer an alternative preventive and therapeutic strategy against HCV infection. DNA vaccination has been shown to induce robust humoral and cellular immunity and overcome many problems associated with conventional vaccines. In this study, mice were primed with either conventional pVRC-based or suicidal pSC-based DNA vaccines carrying DEC-205-targeted NS3 antigen (DEC-NS3) and boosted with type 5 adenoviral vectors encoding the partial NS3 and core antigens (C44P). The prime boost regimen induced a marked increase in antigen-specific humoral and T-cell responses in comparison with either rAd5-based vaccines or DEC-205-targeted DNA immunization in isolation. The protective effect against heterogeneous challenge was correlated with high levels of anti-NS3 IgG and T-cell-mediated immunity against NS3 peptides. Moreover, priming with a suicidal DNA vaccine (pSC-DEC-NS3), which elicited increased TNF-α-producing CD4+ and CD8+ T-cells against NS3-2 peptides (aa 1245–1461), after boosting, showed increased heterogeneous protective potential compared with priming with a conventional DNA vaccine (pVRC-DEC-NS3). In conclusion, a suicidal DNA vector (pSC-DEC-NS3) expressing DEC-205-targeted NS3 combined with boosting using an rAd5-based HCV vaccine (rAd5-C44P) is a good candidate for a safe and effective vaccine against HCV infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis C virus (HCV) infection is a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma, affecting approximately 3 % of the population worldwide [1]. HCV remains a major global health problem due to a lack of affordable preventative vaccines, which remain to be the optimal long-term strategy to control this global epidemic [2]. Currently, the treatment for HCV is expensive, and its efficacy is limited by specific HCV genotypes, adverse effects, comorbidities (e.g., cirrhosis or HIV co-infection) and host factors [2, 3], although the development of direct-acting antivirals (DAAs) will markedly improve the outcome of antiviral treatment [4, 5]. Therefore, there is an urgent need for alternative therapies and effective prophylactic and therapeutic vaccines [2, 6].
Considering the high genetic diversity of HCV [1, 7], a successful vaccine will need to induce potent, multi-specific, and long-lasting T-cell responses that can protect against diverse HCV genotypes and inhibit cell-to-cell viral transmission [6, 8]. The non-structural protein 3 (NS3) of HCV carries a variety of CD4+ and CD8+ T-cell epitopes and induces strong HCV-specific T-cell responses [9], which are correlated with viral clearance and resolution of acute HCV infection. Furthermore, the HCV core protein carries a variety of CD4+ and CD8+ epitopes [10]. Therefore, from an antigenic perspective, NS3 and the core protein represent attractive candidates for use in therapeutic vaccines eliciting T-cell-mediated immune responses against HCV [6, 9–11].
A practical advantage of DNA vaccines is that they can be easily produced in large quantities under standardized conditions compared to recombinant protein vaccines, which require adjuvant for each antigen [12, 13]. DNA-based alphaviral RNA replicon vectors, also known as suicidal DNA vectors [14, 15], have been employed because they are safer than conventional DNA vaccines, which, for example, have the potential to integrated into the host genome and cause cell transformation [13–15]. The use of suicidal vectors is particularly desirable for the development of vaccines targeting potentially oncogenic proteins [15], such as the NS3 protein of HCV. The immunogenicity of DNA vaccines could be enhanced by targeting the encoded protein using DEC-205-expressing dendritic cells (DCs) [16–18]. Our previous data also indicated that immunizing mice, using in vivo electroporation with DNA vaccines encoding HCV NS3 fused to single-chain antibodies to DEC-205, enhances T-cell immunity to a greater degree compared with non-targeted DNA vaccination [19]. In addition, adenovirus (Ad) vectors are effective tools for inducing T-cell immunity against pathogens in animal and humans [20–24]. We previously reported that adenoviral vectors expressing the HCV E1E2 protein induce protective immune responses in mice [23]. Furthermore, the immunogenicity of viral vector vaccines is improved demonstrably by priming with DNA vaccines in various animal models [25, 26], which clearly demonstrates the utility of this vaccine regimen in the development of HCV vaccine. Further studies assessing the protective potential of combining this approach with optimized immunogens and vaccination regimes are warranted [6, 25–27].
In the present study, we evaluated two different DNA-backbone-based vaccines (i.e., pVRC- and pSC-DNA vectors) expressing DEC-205 and full-length NS3 of HCV, in isolation and in a prime-boost vaccination using recombinant human serotype 5 Ad vectors (Ad5) expressing truncated NS3 and core fusion proteins of HCV[24]. This combined regimen induced potent T-cell and high-titre functional IgG responses in mice. Importantly, immunity was protective against heterogeneous HCV strains in a surrogate challenge model [23, 28, 29].
Materials and methods
Preparation of DNA- and recombinant adenoviral-vector-based HCV vaccines
The pVRC vector was provided by Dr. Gary Nabel, and was described previously [30]. In this DNA vector, optimization of transcriptional regulatory elements was applied to the conventional plasmid backbone (the regulatory R element from HTLV-1 to a standard CMV promoter/enhancer) to improve antigen expression and augment the vaccine-elicited cellular immune responses induced by DNA vaccines.
The pSC vector (kindly donated by Dr. Rod Bremner of the University of Toronto) contains the human cytomegalovirus immediate-early gene (HCMV IE) promoter, upstream of the Semliki Forest virus replicon [15]. The subgenomic promoter is located downstream of the Semliki Forest virus replicon, upstream of a multiple cloning site for the insertion of genes of interest.
To target the plasmid-encoded antigens to DC, they were fused to a single-chain antibody directed against the DC surface receptor DEC205 as described previously [19]. The fused gene, comprising the DEC-205 and HCV NS3 genes (aa 1027-1657, HCV 1b serotype), was inserted into the conventional DNA vector pVRC and the suicidal DNA vector pSC, named pVRC-DEC-NS3 and pSC-DEC-NS3, respectively (Fig. 1A).

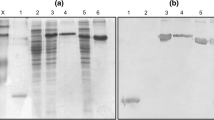
Production and validation of HCV vaccine candidates. A) Schematic representation of two HCV DNA vaccine candidates and recombinant adenovirus rAd5-C44P. ITR, inverted terminal repeat. B) HCV immunogen expression of HCV DNA and rAd vaccine candidates, as detected by Western blot. The DEC-NS3 fusion gene comprises the antibody to DEC-205 (285 aa) and the HCV NS3 gene (631 aa). The C44P gene contains part of the NS3 protein (265 aa) and part of the core protein (34 aa). The bands corresponding to the NS3 protein are indicated
The recombinant adenoviral-based vaccine (rAd5-C44P), carrying the hepatitis C virus truncated NS3 (1201-1465 aa) and core (1-34 aa) fusion protein, was constructed as described previously [24].
Characterization of the immunogens
Expression of the target protein was evaluated by Western blot analysis, using a mouse monoclonal antibody (mAb) against HCV NS3. 293FT cells transfected with the DNA vectors or infected with rAd5-C44P (MOI = 0.1) were harvested after 48 h. Cell lysates were then separated on a 10 % or 15 % polyacrylamide gel and transferred by electroblotting to a polyvinylidene fluoride (PVDF) membrane. The membrane was incubated with a mAb against HCV NS3 (purchased from DAKO) overnight at 4 °C, followed by incubation with goat anti-mouse antibody (IRDye 800) for 1 h at 37 °C. Contact with light was avoided. The band of interest was analysed using an infrared imaging system.
Immunization and challenge of mice
Animal studies were carried out in strict compliance with the Guide for the Care and Use of Laboratory Animals of the People’s Republic of China. The study protocol was approved by the Committee on the Ethics of Animal Experiments of the Chinese Centre for Diseases Control and Prevention. All procedures were performed under ethyl ether anesthesia, and all efforts were made to minimize suffering.
Groups of female BALB/c mice were immunized between 6 and 8 weeks of age (Table 1). Mice were immunized twice with pVRC-DEC-NS3 and pSC-DEC-NS3 administered by intradermal injection combined with electroporation [31, 32], and subsequently boosted by intramuscular injection with rAd5-C44P at 3-week intervals (Fig. 2). Mice immunized with PBS were used as controls. Three weeks after the final immunization, mice in the immunized and PBS control groups were challenged by intraperitoneal injection (i.p.) with 1 × 107 pfu of rTTV-JFH1 (full ORF, 1-3011 aa, 2a genotype JFH1 strain) [23]. Mice were killed, and their ovaries were harvested 5 days after challenge (i.e., at the peak of vaccinia virus titre in the ovary). After freeze-thawing and homogenisation, the vaccinia virus titre was determined on chicken embryo cells by plaque assay.

Combined immunization using HCV DNA and rAd vaccine candidates. Mice were immunized with HCV DNA vaccine candidates or recombinant adenovirus at 3-week intervals. Antibody and cellular immune responses were assessed 2 weeks after prime or boost. Mice were challenged with rTTV-JFH1 at week 10. The protective effects of immunization were evaluated by analysing the vaccinia virus titre in mouse ovaries 5 days post-challenge
Analysis of the immune response in vaccinated mice
Synthetic peptides
A peptide library of HCV non-structural protein NS3 (Table S1), based on the Hebei strain sequence of HCV genotype 1b, was synthesized with 13-17 amino acids (aa), with a 10-aa overlap with the preceding peptides (ZhongKeYaGuan Co., Beijing, China). The peptides were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 50 mg/ml. The NS3 peptide pool (containing 86 peptides) produces the NS3-1 (30 peptides, covering aa 1026-1244), NS3-2 (30 peptides, covering aa 1245-1461) and NS3-3 (26 peptides, covering aa 1462-1657) pools. Peptide pools were aliquoted, stored at −20 °C and used at a final concentration of 4 µg/mL.
Enzyme-linked immunospot (ELISPOT) assays
Splenocytes were harvested 2 weeks after immunization and stimulated using the HCV peptide pools. Briefly, each well of a multiscreen 96-well plates (BD Pharmingen) was coated overnight with 100 μl of 5 μg/ml anti-human gamma interferon antibody (IFN-γ; BD Pharmingen) in PBS. The plates were then washed three times with RPMI 1640 containing 10 % FBS, blocked for 2 h with RPMI 1640 containing 10 % FBS, and incubated with peptide pools and mononuclear spleen cells (MNCs) in triplicate in 100-μl reaction mixture volumes. Each peptide was present at 4 μg/ml in the pools. The reaction was performed as described previously [23]; spots were analysed automatically using an ELISPOT plate reader (Bioreader).
Intracellular cytokine staining (ICS)
Splenocytes (2 × 106/sample) were cultured for 5 h at 37 °C in a 96-well round-bottom microtiter plate in RPMI 1640 supplemented with 10 % FBS and peptide pools and brefeldin A were then added and incubated with the cells for 12 h before staining. Peptides in pools were used at a concentration of 4 µg/ml to stimulate the cells. Control cells were incubated with an unrelated peptide or without peptide. After washing, cells were incubated for 30 min at 4 °C with 25 µl of a 1/100 dilution of an FITC-labelled antibody targeting mouse CD8 and PerCP-labelled CD4. They were washed again and permeabilised in 1 × Cytofix/Cytoperm for 20 min at 4 °C, washed three times with Perm/Wash, and incubated in the same buffer for 30 min at 4 °C with 25 µl of a 1/100 dilution of APC-labelled antibody targeting mouse IFN-γ or TNF-α and of PE-labelled antibody targeting mouse IL-2 or IL-4. After washing, cells were examined by three-color flow cytometry. The above reagents and antibodies were purchased from BD PharMingen (San Diego, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
Two weeks after the second injection with PBS or DNA, blood was collected from the animals and sera were applied to a 96-well plate pre-coated with C44P (containing partial core and NS3) protein for detection of the antibody response as described previously [19, 24]. The antibody titre was defined as the reciprocal of the highest serum dilution giving a positive result.
Data analysis
Significant differences between the experimental and control groups were evaluated by one-way ANOVA, performed using the SPSS for Windows software package (ver. 12.1; SPSS Inc., Chicago, IL, USA). A p-value <0.05 was taken to indicate statistical significance.
Results
Characterization of DNA vectors
Schematic diagrams of both the DNA and adenoviral-based vaccines used in this study are shown in Figure 1A. Target protein expression of the HCV vaccine was confirmed by Western blot analysis (Fig. 1B). Normalization was done using actin protein. The level of target protein expression in cells transfected with pVRC vector was higher than with the pSC vector, and bands corresponding to certain truncated or cleaved proteins were also detected in samples from cells transfected with DNA. We suggest that the bands between 75 and 100 kDa are degradation fragments of DEC-NS3.
Higher antibody levels in mice primed with the conventional DNA vector than in those primed with suicidal DNA vector
To compare humoral responses among groups, the antibody titres of mouse sera were determined by ELISA using purified C44P antigen 2 weeks after injection. After two injections with PBS or DNA, the IgG titres induced by the conventional non-replicating DNA vector pVRC-DEC-NS3 were higher than that induced by suicidal DNA vector pSC-DEC-NS3 (p < 0.05). After boosting with rAd5-C44P, the IgG antibody titres in each group increased significantly. Similarly, the IgG antibody titres in the pVRC-DEC-NS3-primed group were higher than in the other groups (p < 0.05), and the DNA-primed groups had markedly higher titres than those receiving rAd vector immunization alone (Fig. 3).

Anti-C44P IgG titres induced by the various HCV vaccine candidates. Sera were collected 2 weeks after the second prime or boost immunization. The antibody titre was defined as the reciprocal of the highest serum dilution that gave a positive result. The data are represented as the mean ± SD from each group. *, p < 0.05; **, p < 0.01
Induction of cellular immune responses in mice immunized with DNA vectors or DNA combined with the rAd vector
To compare the cellular immune responses elicited by pVRC-DEC-NS3 and pSC-DEC-NS3 priming, ELISPOT was used to determine the number of cells secreting IFN-γ 2 weeks after immunization (Fig. 4). Splenocytes were harvested and stimulated with HCV peptide pools representing NS3 protein. Spot-forming cell (SFC) levels in the pVRC-DEC-NS3-primed group were higher than in the pSC-DEC-NS3-primed group with no statistical significance. The levels of SFC detected subsequent to boosting with the rAd vector increased substantially in the groups. DNA combined with the rAd vector induced a marked increase in cellular immunity compared with rAd vector immunization alone.

NS3-specific cellular immune responses to vaccination with HCV NS3-based DNA and rAd vaccine candidates, detected by ELISPOT. An ELISPOT assay was carried out using splenocytes stimulated with an HCV NS3 peptide pool. Splenocytes were isolated from mice (N = 6 per group) at weeks 5 (prior to boosting) and 8 (subsequent to boosting). The data are expressed as spot-forming cell (SFC) responses to stimulation with the NS3 peptide pool and are presented as the mean (±SEM) of triplicate measurements per mouse. p-values indicative of differences between groups are provided. Control-group mice injected with PBS generated SFC responses of <20/106 mononuclear cells. p-values indicative of differences between groups are provided. *p < 0.05
Intracellular cytokine staining was performed to evaluate the functionality of T-lymphocyte responses in the spleen after the final immunisation (Table 2). A preliminary experiment indicated that the cellular immune responses detected in groups stimulated with the NS3-3 peptide pool was much weaker than the response elicited by the NS3-1 or NS3-2 peptide pool (data not shown). The splenocytes used for ICS in this study were stimulated using the NS3-1 and NS-2 peptide pools. After stimulation with the NS3-1 peptide pool, TNF-α-positive CD8+ (1.2 %) and CD4+ T cells (2.5 %) and IL-2+ CD4+ T-cells (0.9 %), were present at higher levels in the pVRC-DEC-NS3 group compared with the pSC-DEC-NS3-primed group. In contrast, stimulation with the NS3-2 peptide pool was associated with higher levels of TNF-α-secreting CD4+ (0.8 %) and CD8+ T cells (0.5 %) in the pSC-DEC-NS3-primed group compared with the pVRC-DEC-NS3-primed group (in which the levels were close to zero).
Protective immunity in a surrogate challenge model
To assess the protective effects of the immunity induced by DNA and rAd vectors (1b), heterogeneous recombinant vaccinia virus (1 × 107 pfu rTTV-JFH1 (2a)) was used as a surrogate challenge virus and inoculated into each group of mice (except group B) 4 weeks after the final injection. Vaccinia virus titres in mouse ovaries were determined 5 days after challenge. As shown in Figure 5, immunization by DNA-rAd prime-boost resulted in protection from heterologous challenge in both groups. Furthermore, a greater protective effect was observed in the pSC-DEC-NS3 group than in the pVRC-DEC-NS3-primed group.

Vaccinia virus titration after challenge with rTTV-JFH1 in mice vaccinated with a combination of HCV NS3-based DNA and rAd vaccine candidates. Mice (N = 6 per group) were sacrificed 5 days post-challenge. Vaccinia virus titres in ovaries were determined. The data are presented as the means (±SEM) for each group. p-values indicative of differences between groups are provided. *, p < 0.05 **, p < 0.01
Discussion
Recently, strategies for developing a therapeutic vaccine against HCV infection have focused on inducing T-cell responses to NS antigens [6, 10, 11, 22, 25, 33–38]. Ad vectors are also being employed in a phase I vaccine trial to deliver NS HCV proteins (NS3–5B) to 36 healthy volunteers, and many promising vaccine approaches have been evaluated in clinical trials [6, 22, 35]. In addition, despite considerable progress in the development of immunocompetent mouse models using different high-end technologies, most available small-animal models are unsuitable for challenge experiments in HCV vaccine studies. Challenge with HCV-recombinant vaccinia virus in mice is one of broadly acceptable surrogate models for evaluation of HCV vaccination [23, 28, 29]. Therefore, the full-length NS3 coding gene targeting DCs and the partial recombinant NS3 and core coding genes from HCV 1b were used in this study, and a surrogate challenge model was used here to investigate the immunity and protective effects of a combination strategy involving HCV DNA vaccine priming with different vector backbones and boosting with an rAd5-based HCV vaccine. Because it is important in the development of a T-cell-based HCV vaccine to induce cross-protection, we use a recombinant virus carrying the JFH1 strain (2a) instead of an HCV 1b strain to evaluate the cross-protection ability of the HCV 1b-based vaccine. However, in the future, we will explore the heterogeneous protective immunity using a recombinant virus carrying another HCV 1b strain (different from the strain we used here for vaccine construction).
Previous studies by our group have shown that intradermal injection of a DNA vaccine combined with electroporation was more effective than other injection procedures (including intramuscular injection with electroporation or intramuscular injection alone) [31, 32], and we therefore used intradermal injection with electroporation in the present study. Our data show that a conventional pVRC-based DNA vaccine induced higher levels of target protein expression after transfection and antigen-specific antibodies after priming when compared with suicidal pSC-based DNA. These results are in accord with previous studies [14, 15]. It has become increasingly accepted that virus-specific CD4+ and CD8+ T-cell responses play a major role in the outcome of HCV infection. Previous studies have shown that clearance of HCV and protection from reinfection is associated with stronger multifunctional Th1-based T-cell responses to HCV antigens [6, 7, 26]. Therefore, we analyzed the T-cell responses by ELISPOT and ICS of Th1 cytokines (IFN-γ or TNF-α) and Th2 cytokines (IL-4) on CD4+ or CD8+ T cells after in vitro stimulation with overlapping HCV NS3 peptide pools for either NS3-1 or NS3-2. Our data indicate that a robust antigen-specific T-cell response was induced by DNA priming and was further boosted with rAd5-C44P (Fig. 4). In addition, the protective effect of heterogeneous challenge in mice was associated with high levels of anti-NS3 IgG and cytokine-producing CD4+ and CD8+ T cells against the NS3 peptide, induced by either pVRC-DEC-NS3 or pSC-DEC-NS3 in a prime-boost vaccination using rAd5-C44P. Conventional DNA vaccine priming elicited markedly higher levels of anti-NS3 IgG and TNF-α-producing CD4+ and CD8+ T cells against NS3-1 peptides (aa 1026–1244) after boosting. However, priming with a suicidal DNA vaccine (pSC-DEC-NS3), which elicited significant levels of TNF-α-producing CD4+ and CD8+ T-cells against NS3-2 peptides (aa 1245–1461) after boosting, was associated with greater heterogeneous protective potential compared with conventional DNA vaccine (pVRC-DEC-NS3) priming. We aligned the protein sequences of NS3 in DEC-NS3 (Hebei) and rTTV-JFH1 (data not shown). No significant differences were found between DEC-NS3 (Hebei) and JFH1 in the region of aa 1245-1461 (NS3-2 peptides). Because the levels of SFC-secreting IFN-γ detected after boosting did not differ among the DNA-primed groups, we suggest that the pSC-DEC-NS3-priming-induced TNF-α-producing T-cell response against NS3-2 peptides might facilitate HCV clearance. This is consistent with a recent report [33] in which clearance of infection in chimpanzees was associated both with a high percentage of polyfunctional cytokine-producing T cells against NS3 peptides and a strong cytolytic T-cell response against epitope NS3 1258–1272.
Targeting of antigens to DCs via the DEC-205 receptor increases both antigen-derived peptides on MHC-I and MHC-II molecules and antigen-specific immune responses [16–18]. In the present study, the DEC-205-targeted DNA prime adenoviral boost regimen also appeared to induce markedly stronger antigen-specific humoral and T-cell responses compared with previous rAd5-based vaccines and DEC-205-targeted DNA immunization experiments [19, 24].
In conclusion, the present study demonstrated that a suicidal DNA vector (pSC-DEC-NS3) expressing DEC-205-targeted NS3 combined with a boost vaccination with a recombinant rAd5-based HCV vaccine (rAd5-C44P) is a good candidate for a safe and efficient therapeutic vaccine against HCV infection. The limitations of the surrogate models for evaluation of HCV vaccination used here might include the restriction of the analysis of virus titre in the ovaries to single time point and failure to detect immunity in the liver, the major site of natural HCV infection. These recombinant HCV vaccines should be examined in a model against HCV 1b and other genotype. Further validation of the effectiveness of this procedure using the optimal regimen in primate models or humans could provide new hope for the development of effective therapeutic vaccines against HCV.
References
Lavanchy D (2011) Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 17:107–115
Shi C, Ploss A (2013) Hepatitis C virus vaccines in the era of new direct-acting antivirals. Expert Rev Gastroenterol Hepatol 7:171–185
Lange CM, Zeuzem S (2013) Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol 58:583–592
Chatterji U, Garcia-Rivera JA, Baugh J, Gawlik K, Wong KA, Zhong W, Brass CA, Naoumov NV, Gallay PA (2014) The combination of alisporivir plus an NS5A inhibitor provides additive to synergistic anti-hepatitis C virus activity without detectable cross-resistance. Antimicrob Agents Chemother 58(6):3327–3334
Everson GT, Sims KD, Rodriguez-Torres M, Hézode C, Lawitz E, Bourlière M, Loustaud-Ratti V, Rustgi V, Schwartz H, Tatum H, Marcellin P, Pol S, Thuluvath PJ, Eley T, Wang X, Huang SP, McPhee F, Wind-Rotolo M, Chung E, Pasquinelli C, Grasela DM, Gardiner DF (2014) Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology 146(2):420–429
Houghton M (2011) Prospects for prophylactic and therapeutic vaccines against the hepatitis C viruses. Immunol Rev 239:99–108
Neumann-Haefelin C, Thimme R (2013) Adaptive immune responses in hepatitis C virus infection. Curr Top Microbiol Immunol 369:243–262
Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, Barnes E (2014) Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. doi:10.1002/hep.27259
Diepolder HM, Zachoval R, Hoffmann RM, Wierenga EA, Santantonio T, Jung MC, Eichenlaub D, Pape GR (1995) Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 346(8981):1006–1007
Habersetzer F, Baumert TF, Stoll-Keller F (2009) GI-5005, a yeast vector vaccine expressing an NS3-core fusion protein for chronic HCV infection. Curr Opin Mol Ther 11(4):456–462
Mikkelsen M, Holst PJ, Bukh J, Thomsen AR, Christensen JP (2011) Enhanced and sustained CD8+ T cell responses with an adenoviral vector-based hepatitis C virus vaccine encoding NS3 linked to the MHC class II chaperone protein invariant chain. J Immunol 186(4):2355–2364
Abdulhaqq SA, Weiner DB (2008) DNA vaccines: developing new strategies to enhance immune responses. Immunol Res 42(1–3):219–232
Barouch DH (2006) Rational design of gene-based vaccines. J Pathol 208:283–289
Hsu KF, Hung CF, Cheng WF, He L, Slater LA, Ling M, Wu TC (2001) Enhancement of suicidal DNA vaccine potency by linking Mycobacterium tuberculosis heat shock protein 70 to an antigen. Gene Ther 8(5):376–383
Kim TW, Hung CF, Juang J, He L, Hardwick JM, Wu TC (2004) Enhancement of suicidal DNA vaccine potency by delaying suicidal DNA-induced cell death. Gene Ther 11(3):336–342
Boscardin SB, Hafalla JCR, Masilamani RF, Kamphorst AO, Zebroski HA, Rai U, Morrot A, Zavala F, Steinman RM, Nussenzweig RS, Nussenzweig MC (2006) Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J Exp Med 203(3):599–606
Nchinda G, Kuroiwa J, Oks M, Trumpfheller C, Park CG, Huang Y, Hannaman D, Schlesinger SJ, Mizenina O, Nussenzweig MC, Uberla K, Steinman RM (2008) The efficacy of DNA vaccination is enhanced in mice by targeting the encoded protein to dendritic cells. J Clin Invest 118(4):1427–1436
Grossmann C, Tenbusch M, Nchinda G, Temchura V, Nabi G, Stone GW, Kornbluth RS, Uberla K (2009) Enhancement of the priming efficacy of DNA vaccines encoding dendritic cell-targeted antigens by synergistic toll-like receptor ligands. BMC Immunol 10:43
Yin X, Wang W, Tan WJ, Deng Y, Guan J, Wen B, Chen H, Ruan L (2011) Enhancement of the immune response in mice with a noval HCV DNA vaccine targeting NS3 to dendritic cells. Bing Du Xue Bao 27(1):44–49
Draper SJ, Heeney JL (2010) Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbial 8:62–73
Tatsis N, Ertl HC (2004) Adenoviruses as vaccine vectors. Mol Ther 10:616–629
Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, Brown A, Antrobus R, Ammendola V, Naddeo M, O’Hara G, Willberg C, Harrison A, Grazioli F, Esposito ML, Siani L, Traboni C, Oo Y, Adams D, Hill A, Colloca S, Nicosia A, Cortese R, Klenerman P (2012) Novel adenovirus-basedvaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med 4:115ra111
Guan J, Wen B, Deng Y, Zhang K, Chen H, Wu X, Ruan L, Tan W (2011) Effect of route of delivery on heterologous protection against HCV induced by an adenovirus vector carrying HCV structural genes. Virol J 4(8):506
Guan J, Deng Y, Chen H, Yang Y, Wen B, Tan W (2014) Immunogenicity and heterologous protection in mice with a recombinant adenoviral-based vaccine carrying a hepatitis C virus truncated NS3 and core fusion protein. Bing Du Xue Bao (in press)
Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, Siani L, Naddeo M, Grazioli F, Esposito ML, Ambrosio M, Sparacino A, Bartiromo M, Meola A, Smith K, Kurioka A, O’Hara GA, Ewer KJ, Anagnostou N, Bliss C, Hill AV, Traboni C, Klenerman P, Cortese R, Nicosia A (2012) Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med 4:115ra112
Folgori A et al (2006) A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med 12:190–197
Matsui M, Moriya O, Akatsuka T (2003) Enhanced induction of hepatitis C virus-specific cytotoxic T lymphocytes and protective efficacy in mice by DNA vaccination followed by adenovirus boosting in combination with the interleukin-12 expression plasmid. Vaccine 21(15):1629–1639
Arribillaga L, de Cerio AL, Sarobe P, Sarobe P, Casares N, Gorriaiz M, Vales A, Bruna-Romero O, Borras-Cuesta F, Paranhos-Baccala G, Prieto J, Ruiz J, Lasarte JJ (2002) Vaccination with an adenoviral vector encoding hepatitis C virus (HCV) NS3 protein protects against infection with HCV-recombinant vaccinia virus. Vaccine 21(3–4):202–210
Murata K, Lechmann M, Qiao M, Gunji T, Alter HJ, Liang TJ (2003) Immunization with hepatitis C virus-like particles protects mice from recombinant hepatitis C virus-vaccinia infection. Proc Natl Acad Sci USA 100(11):6753–6758
Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, Letvin NL, Nabel GJ (2005) A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J Virol 79:8828–8834
Yin X, Lu J, Tan W (2010) Enhancement of the immune response of a novel DNA vaccine encoding conserved NS3 and Core fusion gene of HCV injected by intradermal electrotransfer in mice. Chin J Microbiol Immunol 30:41–45
Chen H, Wen B, Deng Y, Wang W, Yin X, Guan J, Ruan L, Tan W (2011) Enhanced effect of DNA immunization plus In Vivo electorporation with a combination of Hepatitis B Virus Core-PreS1 and S-PreS1 plasmids. Clin Vaccine Immunol 18:1789–1795
Verstrepen BE, Verschoor EJ, Fagrouch ZC, Mooij P, de Groot NG, Bontrop RE, Bogers WM, Heeney JL, Koopman G (2014) Strong vaccine-induced CD8 T-cell responses have cytolytic function in a chimpanzee clearing HCV infection. PLoS One 9(4):e95103
Ahlén G, Holmström F, Gibbs A, Alheim M, Frelin L (2014) Long-term functional duration of immune responses to HCV NS3/4A induced by DNA vaccination. Gene Ther. 21(8):739–750
Lang Kuhs KA, Ginsberg AA, Yan J, Wiseman RW, Khan AS, Sardesai NY, O’Connor DH, Weiner DB (2012) Hepatitis C virus NS3/NS4A DNA vaccine induces multiepitope T cell responses in rhesus macaques mimicking human immune responses [corrected]. Mol Ther 20(3):669–678
Ip PP, Boerma A, Regts J, Meijerhof T, Wilschut J, Nijman HW, Daemen T (2014) Alphavirus-based vaccines encoding nonstructural proteins of hepatitis C virus induce robust and protective T-cell responses. Mol Ther 22(4):881–890
Fournillier A, Frelin L, Jacquier E, Ahlén G, Brass A, Gerossier E, Holmström F, Broderick KE, Sardesai NY, Bonnefoy JY, Inchauspé G, Sällberg M (2013) A heterologous prime/boost vaccination strategy enhances the immunogenicity of therapeutic vaccines for hepatitis C virus. J Infect Dis 208(6):1008–1019
Acknowledgments
The authors thank Dr. Gary Nabel (VRC, NIH, USA) for providing the pVRC vector, Dr. Rod Bremner (University of Toronto, Canada) for providing the pSC vector and Dr. R.M. Steinman (The Rockefeller University, USA) for providing the DEC205 plasmid. This study was supported by the 863 Hi-Tech Research and Development Program of China (2007AA02Z455) and the National Natural Science Foundation of China (81373229).
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Guan and Y. Deng are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guan, J., Deng, Y., Chen, H. et al. Priming with two DNA vaccines expressing hepatitis C virus NS3 protein targeting dendritic cells elicits superior heterologous protective potential in mice. Arch Virol 160, 2517–2524 (2015). https://doi.org/10.1007/s00705-015-2535-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2535-7