
Abstract
Based on morphology and comparative nucleotide and protein sequence analysis, a new subfamily of the family Siphoviridae is proposed, named “Jerseyvirinae” and consisting of three genera, “Jerseylikevirus”, “Sp3unalikevirus” and “K1glikevirus”. To date, this subfamily consists of 18 phages for which the genomes have been sequenced. Salmonella phages Jersey, vB_SenS_AG11, vB_SenS-Ent1, vB_SenS-Ent2, vB_SenS-Ent3, FSL SP-101, SETP3, SETP7, SETP13, SE2, SS3e and wksl3 form the proposed genus “Jerseylikevirus”. The proposed genus “K1glikevirus” consists of Escherichia phages K1G, K1H, K1ind1, K1ind2 and K1ind3. The proposed genus “Sp3unalikevirus” contains one member so far. Jersey-like phages appear to be widely distributed, as the above phages were isolated in the UK, Canada, the USA and South Korea between 1970 and the present day. The distinguishing features of this subfamily include a distinct siphovirus morphotype, genomes of 40.7-43.6 kb (49.6-51.4 mol % G+C), a syntenic genome organisation, and a high degree of nucleotide sequence identity and shared proteins. All known members of the proposed subfamily are strictly lytic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The advent of affordable whole-genome sequencing and the renewed interest in using bacteriophages as alternatives to antibiotics or to decontaminate food substances has led to a marked increase in submissions of complete phage genomes to public databases such as GenBank. While this process has led to comparative genome analyses and resulted in the recognition of relationships between newly submitted phage genome sequences and those already deposited, official recognition of these new taxonomic units has lagged. This is particularly true for members of the family Siphoviridae (double-stranded dsDNA genome, non-contractile tail), for which the present International Committee on Taxonomy of Viruses (ICTV) only recognizes 10 genera and 31 species (www.ictvonline.org/virusTaxonomy.org). These species account for less than 10 % of the fully sequenced genomes from members of this family, a situation that must be addressed. A significant number of these unclassified Siphoviridae members Salmonella hosts.
Serovars of Salmonella are widespread aetiological agents of food and waterborne diseases in humans and livestock. Salmonella enterica is classified into six subspecies by biochemical tests. Based upon serology of the lipopolysaccharide (O) and flagella (H) antigens, over 2,600 serovars of Salmonella have been described [22]. Not surprisingly, Salmonella phages are also numerous and varied. The morphology of 177 Salmonella phages was reviewed in 2007 [2] (updated in [55]), with most representatives belonging to the order Caudovirales (tailed phages with a dsDNA genome) and its three families, Siphoviridae, Myoviridae and Podoviridae. A small number of phages representing the families Inoviridae, Leviviridae, Microviridae and Tectiviridae have also been documented.
A number of Salmonella phages were originally isolated for the purpose of phage typing. Phage typing represents a useful epidemiological tool whereby strains of a particular serovar may be differentiated into phage types based on susceptibility to a panel of bacteriophages. Phage Jersey was originally isolated and used by Felix and Callow in the development of a typing scheme for Salmonella paratyphi B [1].
Jersey has a morphotype that is recognizable by electron microscopy, with a tail-terminal disk-like structure with six club-shaped spikes [1, 2, 14]. To date, almost all phages examined that exhibit the Jersey-type morphotype have been restricted to Salmonella and Escherichia, the sole exception to this rule being Serratia phage η [15]. No phages with this morphotype have been observed to infect other Gram-negative genera, including Aeromonas, Pseudomonas, Vibrio, or Rhizobium (Table 1). The genome of phage Jersey has recently been sequenced, and discontinuous MEGABLAST analysis revealed similarity to a number of siphovirus genomes deposited in GenBank.
On the basis of an in-depth bioinformatics analysis, this work proposes the creation of three new genera within a novel subfamily, “Jerseyvirinae”.
Results
Morphological characteristics
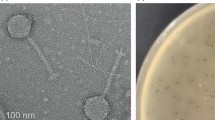
Members of this subfamily that have been examined by transmission electron microscopy are typical siphoviruses with isometric heads of 60 nm in diameter between opposite apices and long, non-contractile tails of 120 × 8 nm (Table 1). Capsids appear as hexagonal or pentagonal and are thus icosahedral in shape. Tails have about 27 transverse striations with a thin, 17-nm-wide baseplate with 5-6 spikes of 10 × 3 nm. Capsid and tail dimensions differ slightly from those of phosphotungstate-stained phage Jersey (head, 68 nm; tail length, 116 nm), which were obtained in 1970 without the benefit of rigorous electron magnification calibration. A morphological diagram of Jersey-like phages is presented in Fig. 1. Phages exhibiting a Jersey-like morphology have been described many times in the literature (Table 1).

Scale drawing of Jersey-like phage (A). Representative negatively stained TEM images of phage Jersey (B) and K1H (C). Scale bars are 100 nm and 20 nm, respectively
Comparative genomics and proteomics of bacteriophages belonging to the proposed subfamily “Jerseyvirinae”
Phages considered in this manuscript are summarised in Table 2. Each was initially linked by similar morphology and discontinuous MEGABLAST analysis.
In addition to phages with completely sequenced genomes, several phages with partial sequences in GenBank were also identified using discontigous MEGABLAST; ST4 [JX233783], L13 [KC832325] and MB78 [AY040866, AF156970, AF349435, AJ277754, AJ249347, AJ245858, AJ245537, X87092, X86562, Y19202, Y19203, Y18133]. These phages are considered tentative members, subject to morphological examination and complete sequencing. Two Salmonella phages, FSL SP-038 and FSL SP-049, are also represented by partial genomic sequences in GenBank. Analyses suggest that they are part of the genus “Sp3unalikevirus”.
Each of the phages listed in Table 2 was colinearized and then examined for overall sequence similarity using progressiveMauve [12] (data not shown), and DNA sequence identity using EMBOSS Stretcher [44] and CLUSTALW [33], which have been widely used for nucleotide sequence alignment of virus genomes [4, 41, 58]. Using the latter methodology, three clades were clearly defined (Fig. 2).

Clustal analysis reveals that the Jersey-like phages fall into three distinct groups, for which we proposed three genera “Jerseylikevirus” (red), “K1glikevirus” (dark blue), and “Sp3unalikevirus” (light blue) within a proposed subfamily, the “Jerseyvirinae.” The genomes were colinearized before analysis. E.coli phage HK578 was included as an outlier. The scale bar represents 0.09 substitutions per site (color figure online)
In addition, the viral proteomes were subjected to pairwise comparisons using CoreGenes 3.0, which provided a measure of the total similarity at the protein level based on pairs of proteins scoring above a pre-defined BLASTP bit score threshold [64]. The CoreGenes and Stretcher results are presented in Table 3. Lastly, using the Markov clustering algorithm OrthoMCL [37] with an e-value threshold of 1e−5 and an inflation value of 1.15, all of the proteins encoded by members of the proposed subfamily were examined (data not shown). Functional annotations of proteins encoded by Jersey-like phages were obtained using BLAST tools and HHpred [50, 51]. Transmembrane domains were identified using TMHMM [31], and conserved domains were identified using Pfam [16] and InterProScan [27]. In line with the 95 % DNA sequence identity threshold used to delineate phage species specified by the Bacterial and Archaeal Viruses Subcommittee of the ICTV, the genus “Jerseylikevirus” comprised 10 species, whose sequence identities range from 71.6 % (FSL SP-101) to 67.6 % (SETP13) and have between 79.7 % and 68.1 % of their proteins in common relative to Jersey (Table 3).
The proposed genus “K1glikevirus” consists of four viruses: K1H, K1G, K1ind1 and K1ind2. They share between 79 % and 97.1 % DNA sequence identity and a minimum of 84 % homologous proteins. Relative to phage Jersey, members of the genus “K1glikevirus” exhibit at minimum 58 % nucleotide sequence identity. Despite exhibiting significant protein homology (66.7 %), the DNA sequence of FSL SP-031 shows only 53.4 % identity to Jersey, a difference sufficient to indicate that this phage is distinct from other members and warrant the creation of a separate genus, “Sp3unalikevirus” within the subfamily.
Based on DNA sequence alignment, protein homology, morphology, and genome organisation, the Jersey-like phages form three distinct groups, united by 53 % DNA sequence identity, and these groups are proposed to represent three genera: “Jerseylikevirus”, “Sp3unalikevirus” and “K1glikevirus” (Fig. 2). In the following sections, the common properties of these viruses, which belong to the proposed subfamily “Jerseyvirinae”, are discussed alongside the specific properties of the members of the three genera.
Protein phylogeny
Phylogenetic trees were constructed to investigate common proteomic features of members of the “Jerseyvirinae” for the large terminase subunit (TerL), portal protein, DNA polymerase (DpoI), helicase, major capsid, and major tail (MTP) proteins (Fig. 3). Analysis of the helicase proteins indicated that this group of phages are phylogenetically related and distinct from the Hk574-like viruses. The trees constructed using the TerL, portal and DpoI sequences clearly indicate that this group of viruses can be subdivided into three clades. Analysis of the major capsid and MTPs reveals that viruses in the K1G clade are significantly different from those in the “Jerseylikevirus” group. Interestingly, the K1H major capsid protein is distinct from those of other members of the subfamily, a feature corroborated by the OrthoMCL groupings.

Phylogenetic analysis of the “Jerseyvirinae” common proteins produced using www.phylogeny.fr. Branch length is proportional to the number of substitutions per site
Common features of members of the “Jerseyvirinae”
The genomes of members of the proposed subfamily “Jerseyvirinae” range in size from 40.7 to 43.6 kb, with a G+C content ranging from 49.6 to 51 %. They also encode between 48 to 69 proteins but no tRNAs. These phages appear to be strictly lytic/virulent, as none have been shown to be capable of lysogeny or to harbour homologues of known integrases, recombinases or excisionases. As with most phages, their genomes exhibit a modular structure, and the organisation of genes shows a high degree of synteny across all members (Fig. 4). The genome may be divided into four modules on the basis of the predicted functions of their component genes: (i) virion structure and assembly, (ii) regulation/immunity, (iii) genome replication and (iv) host lysis. Despite the presence of a number of deduced proteins of unknown function, the roles of some gene products can be predicted on the basis of BLAST and HHpred searches or the presence of conserved domains.

Genetic and physical map of Salmonella phage Jersey prepared using CGView [21]. For sequence similarity comparison, TBLASTX was used versus wks13 (red), FSL-SP031 (dark green) and K1H (dark blue). GC content is depicted in black while positive and negative GC skew is denoted by green and purple, respectively (color figure online)
Using OrthoMCL, the 1,057 proteins encoded by the 18 members of the three proposed genera were found to form 90 clusters of orthologous proteins and 51 singletons. Of these, 25 clusters were present in all members of the three proposed genera, representing conserved proteins in the structural, replicative and lysis gene modules. In addition to these ‘core’ genes/proteins, a further 1, 5 and 12 clusters were present in all members of “Jerseylikevirus”, “K1glikevirus”, and “Sp3unalikevirus”, respectively. The structural gene module represents the most highly conserved region with 17 ortholog clusters. Almost half of the genes encoded by the “Jerseyvirinae” phages are devoted to genome packaging, assembly and structure of mature virions. The structural module follows a strongly conserved gene order, encoding genes involved in DNA packaging, followed by the virion capsid, tail and adsorption apparatus in an arrangement reminiscent of that observed in many phages.
Several features are of interest within the morphogenesis module. Using phage Jersey as reference, the major capsid protein (gp12) shows structural similarity to Bacillus phage SPP1 and coliphage HK97 (Protein Data Bank [PDB] accession numbers: 4an5 and 3p8q, respectively), suggesting that the capsid protein shares the evolutionarily conserved HK97-like fold [17]. Three gene products showed structural similarity to Ig-set domain proteins when analyzed using HHpred. Two of these proteins, Jersey gp7 (AGP24895) and SETP13 gp12 (AGX84616), returned matches to the Hoc protein of enterobacteria phage RB49 (PDB 3shs) while a third, Jersey gp13 (AGP24902), returned a match to the head fibre of Bacillus phage Φ29 (PDB 3qc7). Ig-like domains are found widely in members of the order Caudovirales and are predominantly associated with structural proteins, suggesting that these proteins play a role in capsid assembly or completion and may also be involved in nonspecific binding to host cells [18]. The presence of a weak match to an Ig-like I-set domain (pfam: PF07679) in gp07 and the proximity of its gene to the gene coding for the major capsid protein suggest that this gene might encode a capsid decoration protein [52]. Head decoration proteins have been described in a number of phages, such as L, λ and ES18, and are thought to aid in the stabilisation of the capsid structure [20]. However, to date, none of the structural proteins present in mature Jersey-like virions have been identified using mass spectrometry.
Using OrthoMCL, it was found that two coding sequences positioned immediately upstream of the gene coding for the putative tape measure form a single cluster that is present in all members of the three proposed genera. The positioning of these genes is similar to that of the λ tail assembly chaperones gpG and gpGT. In λ, gpG and the translational frameshift product gpGT interact with the tape measure and major tail proteins and are required for correct tail formation [60–62]. Analysis of the putative gpG and gpGT genes in phage Jersey (AGP24914 and AGP24915) using MFOLD and HPKNOTTER provided evidence for the presence of a stem-loop structure and a pseudoknot, respectively, suggesting that the Jersey-like phages produce a gpGT-like fusion product.
Using PSI-BLAST, it was found that the putative tail fibre (Jersey gp32, AGP24920) shows distant similarity to the central tail fibre gpJ of phage λ and p33 of phage T1 and is conserved among all members of the “Jerseyvirinae”. However, a central tail tip fibre has not been observed in electron micrographs, so the precise role and location of this protein remain unclear. This protein may in fact form the virion baseplate in conjunction with a gene (Jersey gp31, AGP24919) encoded immediately upstream that exhibits similarity to the endolysin of Pseudomonas phage ϕKZ (PBD: 3bkh), suggesting a role in cell wall degradation.
In all members of the “Jerseyvirinae”, the morphogenesis gene module is interrupted after the gene coding for the major tail protein by a cassette of between 1 to 5 genes encoded in the opposite orientation, where no single protein is present in all members of the subfamily. With the exception of phages SS3e and SP-031, all members of the “Jerseyvirinae” encode a putative serine/threonine protease in this region (pfam:PF12850, metallophos_2), which, according to HHpred, is linked to the NinI protein phosphatase in λ (PDB 1g5b).
Four critical units involved in genome replication, a replicative family A DNA polymerase (pfam: PF00476), DEAD-box helicase-primase (pfam: PF13481) and helicase (pfam: PF00176), are also conserved in all members of the proposed subfamily. Analysis of the replication gene cluster using HHpred provides additional evidence for the presence of a Holliday junction resolvase (PDB 1hh1), a helicase subunit (PDB 3h4r), and a helix-destabilising ssDNA-binding protein similar to gp2.5 of phage T7 (PDB 1je5).
Finally, the lysis gene cluster codes for proteins facilitating lysis of the infected host cell, allowing egress of newly formed virions into the surrounding environment. The lysis or late gene module represents an area of significant divergence among the three genera and between individual phages. The module is replete with ORFs of unknown function, and only three gene products, endolysin, holin, and a protein of unknown function are conserved across the subfamily. While the holin formed a single cluster when analyzed using OrthoMCL, the number of predicted transmembrane domains differed between phages. On the basis of predicted transmembrane domains, phages AG11, Jersey, SE2, SETP3, wksl3, Ent2 and SP-031 are presumed to encode class I holins, while Ent1, Ent3, SP-101, SETP7, SETP13 and SS3e [63] encode class II holins. The endolysin belongs to the glycoside hydrolase 24 family (muraminidase, pfam:PF00959). Many of the lysis gene products are shared between only some members of each genus, while others are found between a limited number of representatives of one or more genera, suggesting that this region has been the site of frequent genetic exchange.
Description of individual genera
“Jerseylikevirus”
This proposed genus is named after the first characterized phage of this morphotype, Salmonella phage Jersey [1]. Members of the genus are distinguished by a distinct morphology, a similar genome size among its members, a conserved gene organisation, and the use of a P22-like tailspike to facilitate host recognition and adsorption, the last of which distinguishes them from members of the genera “K1glikevirus” and “Sp3unalikevirus”, as do the additional 25 accessory genes that are shared between members of two or more species. To date, 12 members of this genus, isolated from four different countries, have been fully sequenced and annotated (Table 2). Members of the genus “Jerseylikevirus” have an average G+C content of 49.79 %, slightly lower than the average of 52 % reported for serovars of Salmonella enterica [40].
Host specificity is conferred by six tailspikes, observed as short clubs attached to the tail terminus. Like the tailspikes of podophages P22, Sf6 and HK620, they exhibit a modular design consisting of a conserved N-terminal binding domain and a P22-like C-terminal catalytic domain (data not shown). In phage P22, the tailspike facilitates adsorption to Salmonella O-antigen 12 of the cell surface LPS, which is expressed by members of White-Kauffmann-Le Minor serogroups A, B and D1 [22]. The high degree of conservation of the P22-like domain and catalytic residues, combined with host-range data for SETP3, SETP7, SETP13, vB_SenS-Ent1 and wksl3 [14, 29, 56], suggests that “Jerseylikevirus” members isolated to date are limited to these Salmonella serogroups. However, SS3e appears to be an exception: it has been reported to be capable of lysing enterobacteria other than Salmonella, including E. coli, Enterobacter cloacae, Shigella sonnei, and Serratia marcescens [56].
Several members of the proposed genus “Jerseylikevirus” (Jersey, Ent1, Ent2, Ent3, SE2, wksl3 and SP-101) encode a putative DNA-binding protein containing one or both of the Pfam-family domains ANT (PF03374) and pRha (PF09669). In phage P22, ant encodes an anti-repressor, which inhibits binding of the c2 repressor to the PL and PR operators, enabling the expression of genes necessary for lytic development [9]. The pRha domain represents a family of proteins whose expression is detrimental for lytic growth in the absence of integration host factor function [54].
A gene product with similarity to inner membrane immunity (Imm) proteins, which protect against superinfecting phages [32], is also found in all Jersey-like phages except Jersey and AG11. This gene product contains an Imm_superinfect motif (pfam: PF14373), is predicted to localize at the cytoplasmic membrane (PSORTb), and contains two transmembrane domains (TMHMM). Only the putative immunity protein [AAZ41745] is annotated in SS3e, although this appears to be due to an incomplete genome sequence rather than the absence of further protein-encoding genes in this region.
An interesting feature of members of “Jerseylikevirus” is that intein insertion is evident in the DNA helicase of phages vB_SenS-Ent1, Ent2, Ent3, SETP3 and SETP7, and also within the DNA polymerase of phages FSL SP-101, Ent1, Ent2, Ent3, SE2 and SETP3 (data not shown). Inteins are defined as protein sequences that are embedded within a precursor sequence which, upon translation, catalyzes self-excision from the host polypeptide and ligation of the flanking sequences to yield two stable products: the mature protein (extein) and the intein [47].
In SE2 and SS3e, the DNA polymerase appears to be encoded by more than one gene. For SE2, two gene products, gp05 (AEX56144) and gp06 (AEX56145) have predicted DNA polymerase activity. The large subunit, gp06, is predicted to possess an intein similar to that found in SETP3 and vB_SenS-Ent1. Like SE2, the DNA polymerase of SS3e also appears to be split into two coding sequences (gp41 and gp43), although in this case, the subunits are interrupted by an additional gene (gp42; AAW51247) with a predicted C-terminal HNH endonuclease domain. Notably, all three gene products show a short match to either the N- or C-terminal end of the SETP3 intein. However, re-examination of these coding sequences suggests that the DNA polymerase in SS3e and SE2 has been split due to sequencing or assembly errors.
A total of 15 accessory genes are encoded within the lysis/late gene cassette, of which only three have homologues with predicted functions: a putative protease (9 members), a HNH homing endonuclease (7 members) and a putative RNA-binding protein (2 members).
K1glikevirus
The Escherichia phages K1G, K1H, K1ind1, K1ind2 and K1ind3, comprising the proposed genus “K1glikevirus”are described as K1-dependent or -independent, denoting the requirement for the K1 capsule for productive infection [8]. The GC content of their genome is slightly higher than that of members of “Jerseylikevirus”, ranging between 51.1 and 51.5 %, a value closer to that of their host, E. coli (50.8 %). Moreover, they encode fewer proteins than do members of “Jerseylikevirus”.
Members of “K1glikevirus” possess tailspikes, which are of similar size to those of members of “Jerseylikevirus”. These proteins exhibit similarity to the conserved N-terminal domain identified in members of “Jerseylikevirus” but possess divergent C-terminal domains, indicative of their different host specificity. Tailspikes from the K1-independent phages K1ind1, K1ind2 and K1ind3 exhibit high similarity to the HK620 tailspike [PDB 2 × 6w] and appear to belong to the pectatelysase 3 family (pfam: PF12708). The HK620 tailspike possess endo-N-acetylglucosaminidase activity, which degrades the O-antigen of E. coli serotype O18A1 [6]. In contrast, the tailspike encoded by the K1-dependent phages K1G and K1H exhibit endo-N-acyl-neuraminidase (endosialidase) activity [8] and are nearly identical to the tailspike of the T7-like phage K1F (PDB 3ju4). Endosialidases bind to and degrade the K1 capsular polysaccharide, a homopolymer made up of α2,8-linked sialic acid residues [6]. Each of the K1-dependent tailspikes is predicted to contain a C-terminal Peptidase_S74 domain (PF13884), which shows homology to the protease domains of K1F, K1E, K1-5 tailspikes as well as the long tail fibre of T5. In these phages, this domain has been shown to function as an intramolecular chaperone, whose presence and subsequent auto-cleavage is essential for folding and assembly of mature proteins [49]. These data indicate that the K1-dependent tailspikes undergo a maturation process that is distinct from those of their counterparts in the genus “Jerseylikevirus” and K1-independent members of the genus “K1glikevirus”.
With the exception of the tailspike proteins and capsid-associated genes in K1H, members of “K1glikevirus” share the complete structural module with phages of the genus “Jerseylikevirus”. Phage K1ind3 has a HNH homing endonuclease gene immediately downstream of a gene coding for a gp7 family morphogenesis protein (gp04) related to Bacillus phage SPO1 (PBD 1u3e). K1G encodes another homing endonuclease (gp12) with 31 % identity to MobE of coliphage T6. Notably, the K1H major capsid sequence differs substantially from those of other “Jerseyvirinae” phages, but it is predicted to have a similar structure to SPP1 and HK97, according to HHpred. A further gene product of unknown function, K1H gp09, which was not found in other members of the proposed subfamily, is encoded immediately downstream of the gene coding for the major capsid protein (K1H gp08, ADA82303).
Three gene products are conserved in all K1G-like phages in the immunity gene module: a serine/threonine protein phosphatase (pfam:PF12850), an acid-phosphatase B domain protein (pfam: PF03767), and a protein predicted to have ATPase activity (pfam: P13207). With the exception of K1H, all members of “K1glikevirus” encode a superinfection immunity protein. Neither Jersey, AG11 nor K1H appear to harbour an immunity protein; instead, these phages encode a hypothetical protein forming a single OrthoMCL cluster.
In addition to the core replication proteins of members of the “Jerseyvirinae”, each of the K1G-like phages encodes a C-5 cytosine-specific methylase (pfam:PF00145) that bears little sequence similarity to the methylase of phage Jersey or FSL SP-101 and was grouped into a separate cluster using OrthoMCL. All K1G-like phages except K1ind1 encode a putative UvsX-like protein. In coliphageT4, UvsX functions as a RecA-like recombinase, interacting with the UvsY helicase to promote strand-exchange during genome replication [19].
Finally, a total of seven proteins are conserved in all K1G-like phages in the lysis or late gene module, three of which are conserved across the subfamily: holin, lysin and a hypothetical protein. Unlike the members of “Jerseylikevirus”, each of the K1G-like phages is predicted to encode a separate class I and class II holin, with three and two transmembrane domains, respectively. Each K1G-like phage has a NinH-like domain protein (pfam:PF06322) in addition to two gene products of unknown function. Lastly, a gene product found only in the K1-independent phages contains a DUF3850 family motif (pfam:PF12961), which has been suggested to be involved in RNA recognition.
“Sp3unalikevirus”
This genus was recently proposed by Moreno Switt et al. (2014). To date, three members (FSL SP-031, FSL SP-038, and FSL SP-049) have been reported, though only the genome of phage FLS SP-031 is fully sequenced, hence the proposed genus “Sp3unalikevirus” [42]. Phages in the genus “Sp03unalikevirus” were isolated from dairy farms in the state of New York with a history of Salmonella isolation [43]. Sp031-like viruses have a GC content of 51.1 %, closer to the Salmonella G+C value of approximately 52 % [46].
The members of “Sp3unalikevirus” are distinguished from members of the other genera by the presence of a number of genes encoding hypothetical products and genes associated with host specificity. The genes coding for a number of unique hypothetical proteins with unknown function are located in the replication module of SP-031-like viruses. Two annotated proteins that are absent in the “Jerseylikevirus” and “K1glikevirus” phages are homing endonucleases. These homing endonucleases are inserted in the replication module of FLS SP-031. One endonuclease shows 41 % identity over 93 % of the protein to an endonuclease of Synechococcus phage S-SSM7 (GenBankYP_004324330.1), and the other endonuclease, with the homing endonuclease domain PF13392, showed 42 % identity over 89 % of the protein to an endonuclease in the genome of Salmonella phage FLS SP-126 (GenBankAGF87903.1). Another difference of Sp03una-like viruses is the presence of a gene encoding a putative phosphoadenosinephosphosulfate (PAPS) reductase (pfam: PF01507) in the virion assembly module. PAPS reductases are involved in the reduction of sulfate to hydrogen sulfide, which is generated by Salmonella in the gut of humans and animals [59]. While PAPS reductases have previously been reported in temperate phages (e.g., BcepB1A [53]), there is no evidence that this enzyme confers a fitness advantage to the host.
Sp031-like phages have a very narrow host range. When 23 different Salmonella serovars were tested, only Salmonella Cerro was lysed [43]. This host specificity is probably determined by the tail spikes in members of “Sp3unalikevirus”. A BLAST search of the tail spike amino acid sequence of FSL SP-031 showed as the best hit (78 % over 80 % of the protein) a tail spike of a P22-like prophage found in the genome of S. Cerro str. 818 (GenBank ESH26034), with divergence at the N-terminal end. This prophage shows no further sequence identity to any other CDSs of Sp031-like phages. To investigate the potential genetic basis of this host range, an alignment of the tail spike amino acid sequences of representative Jersey-like viruses (i.e., Jersey, L3, SETP7, SETP3, SE2, Ent2, SETP12, and FSL SP-101) and Sp031-like viruses (FSL SP-031) showed a conserved N-terminus (approx. 87 % aa identity) and a divergent C-terminus (approx. 15 % aa identity). This finding corresponds to the host specificity reported. While other Salmonella phages in the subfamily “Jerseyvirinae” infect serogroups A, B, and D; Sp031-like viruses only infect serogroup K.
Discussion
The availability of an increasing number of genome sequences and improvements in gene prediction methods has resulted in a sizeable shift towards the inclusion of genomic and proteomic data for the taxonomic classification of bacteriophages [36]. However, a number of different, and sometimes conflicting, approaches have been reported in the literature for the purpose of delineating evolutionary relationships between phages, including the use of proteomic trees [48], analysis of shared homologous/orthologous proteins [34, 35], and reticulate classification based on gene content [38, 39].
Our analysis revealed that the classification criteria introduced for members of the families Podoviridae and Myoviridae [34, 35], based upon the existence of protein homologs, tend to “lump” taxa rather than represent the true taxonomic relationship within a genus. Using an alternative approach, employing NCBI BLASTN and TBLASTX, a genus can be defined as a group in which members share ≥65 % DNA sequence identity, while members of the same subfamily should show ≥40 % protein homologs (Kropinski, Edwards and Mahadevan, unpublished results). These values are those derived by Niu et al. in their assessment of the T1-likeviruses [45].
Phylogenetic analysis also demonstrates that the Salmonella and Escherichia phages described here fall into three clusters, substantiating the establishment of three genera. Analysis using EMBOSS Stretcher demonstrated that while phages within each genera were closely related, a significant relationship based on shared homologous proteins identified using CoreGenes and OrthoMCL existed between genera. Based upon these data, we propose the establishment of the subfamily “Jerseyvirinae” within the family Siphoviridae, comprising three genera, “Jerseylikevirus”, “K1Glikevirus” and “Sp3unalikevirus”. The three genera of closely related phages were isolated from geographically disparate locations. This suggests that phages of this subfamily are widely distributed and have persisted and evolved in various environments. Considering the length of time between the isolation of Jersey and the other members, horizontal gene transfer does not always mask the identification of taxonomic relationships between phages. Grose and Casjens have recently clustered 337 phages infecting various members of family Enterobacteriaceae [23] based upon average nucleotide sequence identity and conserved gene product content in addition to whole-genome nucleotide and amino acid dotplots. They report a “SETP3 supercluster”, formed from five clusters of lytic phages: SETP3-like, SO-1-like, ECO1230-10-like, Gj1-like and PY100-like phages. Our independent findings are in broad agreement with those reported by Grose and Casjens but provide a more detailed analysis of the phages comprising the SETP3-like subcluster.
All Jersey-like phages isolated to date exhibit a strictly lytic lifestyle and encode no proteins with homology to known toxins or allergens. As such, members of this subfamily appear suited to biocontrol applications [7, 24], particularly members of “Jerseylikevirus”, which exhibit a broad host range encompassing important serogroups of Salmonella.
Undoubtedly, more Jersey-like bacteriophages will be isolated in the future. The authors hope the data provided here will act as a starting point for the annotation of these future isolates.
References
Ackermann H-W, Berthiaume L, Kasatiya SS (1972) Morphologie des phages de lysotypie de Salmonella paratyphi B (schéma de Felix et Callow). Can J Microbiol 18:77–81
Ackermann HW, Gershman M (1992) Morphology of phages of a general Salmonella typing set. Res Virol 143:303–310
Ackermann HW, DuBow MS, Gershman M, Karska-Wysocki B, Kasatiya SS, Loessner MJ, Mamet-Bratley MD, Regué M (1997) Taxonomic changes in tailed phages of enterobacteria. Arch Virol 142:1381–1390
Alonso C, Murtaugh MP, Dee SA, Davies PR (2013) Epidemiological study of air filtration systems for preventing PRRSV infection in large sow herds. Prev Vet Med 112:109–117
Anany H (2010) Biocontrol of foodborne bacterial pathogens using immobilized bacteriophages. Food Science. University of Guelph, Guelph
Barbirz S, Muller JJ, Uetrecht C, Clark AJ, Heinemann U, Seckler R (2008) Crystal structure of Escherichia coli phage HK620 tailspike: podoviral tailspike endoglycosidase modules are evolutionarily related. Mol Microbiol 69:303–316
Brovko LY, Anany H, Griffiths MW (2012) Bacteriophages for detection and control of bacterial pathogens in food and food-processing environment. Adv Food Nutr Res 67:241–288
Bull JJ, Vimr ER, Molineux IJ (2010) A tale of tails: sialidase is key to success in a model of phage therapy against K1-capsulated Escherichia coli. Virology 398:79–86
Byl CV, Kropinski AM (2000) Sequence of the genome of Salmonella bacteriophage P22. J Bacteriol 182:6472–6481
Carey-Smith GV, Billington C, Cornelius AJ, Hudson JA, Heinemann JA (2006) Isolation and characterization of bacteriophages infecting Salmonella spp. FEMS Microbiol Lett 258:182–186
Chibani-Chennoufi S, Sidoti J, Bruttin A, Dillmann M-L, Kutter E, Qadri F, Sarker SA, Brüssow H (2004) Isolation of Escherichia coli bacteriophages from the stool of pediatric diarrhea patients in Bangladesh. J Bacteriol 186:8287–8294
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome Alignment with gene gain, loss and rearrangement. PLoS One 5:e11147
De Lappe N, Doran G, O’Connor J, O’Hare C, Cormican M (2009) Characterization of bacteriophages used in the Salmonella enterica serovar Enteritidis phage-typing scheme. J Med Microbiol 58:86–93
Demczuk W, Ahmed R, Ackermann H-W (2004) Morphology of Salmonella enterica serovar Heidelberg typing phages. Can J Microbiol 50:873–875
Denyes J, Krell P, Manderville R, Ackermann H-W, She Y-M, Kropinski A (2014) The genome and proteome of Serratia bacteriophage eta which forms unstable lysogens. Virol J 11:6
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230
Fokine A, Leiman PG, Shneider MM, Ahvazi B, Boeshans KM, Steven AC, Black LW, Mesyanzhinov VV, Rossmann MG (2005) Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc Natl Acad Sci USA 102:7163–7168
Fraser JS, Yu Z, Maxwell KL, Davidson AR (2006) Ig-like domains on bacteriophages: a tale of promiscuity and deceit. J Mol Biol 359:496–507
Gajewski S, Webb MR, Galkin V, Egelman EH, Kreuzer KN, White SW (2011) Crystal structure of the phage T4 recombinase UvsX and its functional interaction with the T4 SF2 helicase UvsW. J Mol Biol 405:65–76
Gilcrease EB, Winn-Stapley DA, Hewitt FC, Joss L, Casjens SR (2005) nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization. J Bacteriol 187:2050–2057
Grant J, Arantes A, Stothard P (2012) Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 13:202
Grimont PAD, Weill F-X (2007) Antigenic formulae of the Salmonella serovars. WHO Collaborating Centre for Reference and Research on Salmonella, Paris, France, p 166
Grose JH, Casjens SR (2014) Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468–470:421–443
Hagens S, Loessner MJ (2010) Bacteriophage for biocontrol of foodborne pathogens: calculations and considerations. Curr Pharm Biotechnol 11:58–68
Hong S, Jeong J, Lee J, Kim S, Min W, Myung H (2013) Therapeutic effects of bacteriophages against Salmonella gallinarum infection in chickens. J Microbiol Biotechnol 23:1478–1483
Hooton SPT, Atterbury RJ, Connerton IF (2011) Application of a bacteriophage cocktail to reduce Salmonella Typhimurium U288 contamination on pig skin. Int J Food Microbiol 151:157–163
Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240
Joshi A, Siddiqi JZ, Rao GR, Chakravorty M (1982) MB78, a virulent bacteriophage of Salmonella typhimurium. J Virol 41:1038–1043
Kang H-W, Kim J-W, Jung T-S, Woo G-J (2013) wksl3, a New biocontrol agent for Salmonella enterica serovars enteritidis and typhimurium in foods: characterization, application, sequence analysis, and oral acute toxicity study. Appl Environ Microbiol 79:1956–1968
Kim S-H, Park J-H, Lee B-K, Kwon H-J, Shin J-H, Kim J, Kim S (2012) Complete genome sequence of Salmonella bacteriophage SS3e. J Virol 86:10253–10254
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Labrie SJ, Samson JE, Moineau S (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8:317–327
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lavigne R, Seto D, Mahadevan P, Ackermann H-W, Kropinski AM (2008) Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol 159:406–414
Lavigne R, Darius P, Summer E, Seto D, Mahadevan P, Nilsson A, Ackermann H, Kropinski A (2009) Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol 9:224
Lawrence JG, Hatfull GF, Hendrix RW (2002) Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol 184:4891–4905
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189
Lima-Mendez G, Van Helden J, Toussaint A, Leplae R (2008) Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol 25:762–777
Lima-Mendez G, Toussaint A, Leplae R (2011) A modular view of the bacteriophage genomic space: identification of host and lifestyle marker modules. Res Microbiol 162:737–746
McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856
Mollov D, Lockhart B, Zlesak D (2013) Complete nucleotide sequence of rose yellow mosaic virus, a novel member of the family Potyviridae. Arch Virol 158:1917–1923
Moreno Switt A, Orsi R, den Bakker H, Vongkamjan K, Altier C, Wiedmann M (2013) Genomic characterization provides new insight into Salmonella phage diversity. BMC Genomics 14:481
Moreno Switt AI, den Bakker HC, Vongkamjan K, Hoelzer K, Warnick LD, Cummings KJ, Wiedmann M (2013) Salmonella bacteriophage diversity reflects host diversity on dairy farms. Food Microbiol 36:275–285
Myers EW, Miller W (1988) Optimal alignments in linear space. Computer applications in the biosciences : CABIOS 4:11–17
Niu YD, McAllister TA, Nash JH, Kropinski AM, Stanford K (2014) Four Escherichia coli O157: H7 phages: a new bacteriophage genus and taxonomic classification of T1-like phages. PLoS One 9:e100426
Papanikolaou N, Trachana K, Theodosiou T, Promponas V, Iliopoulos I (2009) Gene socialization: gene order, GC content and gene silencing in Salmonella. BMC Genomics 10:597
Perler FB, Olsen GJ, Adam E (1997) Compilation and analysis of intein sequences. Nucleic Acids Res 25:1087–1093
Rohwer F, Edwards R (2002) The phage proteomic tree: a genome-based taxonomy for phage. J Bacteriol 184:4529–4535
Schwarzer D, Stummeyer K, Gerardy-Schahn R, Muhlenhoff M (2007) Characterization of a novel intramolecular chaperone domain conserved in endosialidases and other bacteriophage tail spike and fiber proteins. J Biol Chem 282:2821–2831
Söding J (2005) Protein homology detection by HMM–HMM comparison. Bioinformatics 21:951–960
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248
Spinelli S, Bebeacua C, Orlov I, Tremblay D, Klaholz BP, Moineau S, Cambillau C (2014) Cryo-electron microscopy structure of lactococcal siphophage 1358 virion. J Virol 88:8900–8910
Summer EJ, Gill JJ, Upton C, Gonzalez CF, Young R (2007) Role of phages in the pathogenesis of Burkholderia, or ‘where are the toxin genes in Burkholderia phages?’. Curr Opin Microbiol 10:410–417
Susskind MM, Botstein D (1975) Mechanism of action of Salmonella phage P22 antirepressor. J Mol Biol 98:413–424
Switt AIM, Sulakvelidze A, Wiedmann M, Kropinski AM, Wishart DS, Poppe C, Liang Y (2014) Salmonella phages and prophages—genomics, taxonomy and applied aspects. In: Schatten H, Eisenstark A (eds) Salmonella: methods and protocols (methods in molecular biology). Humana Press, New York
Tiwari BR, Kim S, Kim J (2012) Complete genomic sequence of Salmonella enterica serovar Enteritidis phage SE2. J Virol 86:7712
Turner D, Hezwani M, Nelson S, Salisbury V, Reynolds D (2012) Characterization of the Salmonella bacteriophage vB_SenS-Ent1. J Gen Virol 93:2046–2056
Westover KM, Rusinko JP, Hoin J, Neal M (2013) Rogue taxa phenomenon: a biological companion to simulation analysis. Mol Phylogenet Evol 69:1–3
Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ (2010) Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429
Xu J, Hendrix RW, Duda RL (2004) Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell 16:11–21
Xu J, Hendrix RW, Duda RL (2013) A balanced ratio of proteins from gene G and frameshift-extended gene GT is required for phage lambda tail assembly. J Mol Biol 425:3476–3487
Xu J, Hendrix RW, Duda RL (2014) Chaperone-protein interactions that mediate assembly of the bacteriophage lambda tail to the correct length. J Mol Biol 426:1004–1018
Young R, Bläsi U (1995) Holins: form and function in bacteriophage lysis. FEMS Microbiol Rev 17:195–205
Zafar N, Mazumder R, Seto D (2002) CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics 3:12
Acknowledgments
SM acknowledges funding from NSERC of Canada (Discovery Program). SM holds a Tier 1 Canada Research Chair in Bacteriophages. HA and MWG acknowledge funding from Sentinel Bioactive Paper Network.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Anany, H., Switt, A.I.M., De Lappe, N. et al. A proposed new bacteriophage subfamily: “Jerseyvirinae”. Arch Virol 160, 1021–1033 (2015). https://doi.org/10.1007/s00705-015-2344-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2344-z