
Abstract
Coenzyme Q2, polyprenyltransferase (COQ2) variants have been reported to be associated with multiple system atrophy (MSA). However, the relationship between COQ2 variants and familial Parkinson’s disease (PD) remains unclear. We investigated the frequency of COQ2 variants and clinical symptoms among familial PD and MSA. We screened COQ2 using the Sanger method in 123 patients with familial PD, 52 patients with sporadic PD, and 39 patients with clinically diagnosed MSA. Clinical information was collected from medical records for the patients with COQ2 variants. Allele frequencies of detected rare non-synonymous variants were compared by public database of the Exome Aggregation Consortium (ExAC) and Japanese genetic variation database, using Fisher’s exact test. We detected two probands with rare variants in COQ2, the p.P157S from Family A, whose patient was clinically diagnosed as having juvenile PD, and the p.H15 N/p.G331S from Family B, whose patients shared common symptoms of PD. Furthermore, in an association study comparing these familial PD and MSA cases with a public variant database, eight non synonymous variants were detected in COQ2. Three of these were very rare variants, namely, p.P157S, p.L261Qfs*4, and p.G331S, and one variant, p.G21S, was found to show a significant association with familial PD. COQ2 variants rarely may associate with the disease onset of familial PD. Our findings contribute to an understanding of COQ2 variants in neurodegenerative disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple system atrophy (MSA) is an adult-onset neurodegenerative disorder characterized by Parkinsonian features, cerebellar ataxia, autonomic failure, and corticospinal disorders (Fanciulli and Wenning 2015). Given that multiplex familial cases are rarely reported, MSA was thought to be a non-genetic disorder (Hara et al. 2007). However, MSA reportedly involves parkinsonism more often in first-degree relatives compared with controls (Vidal et al. 2010; Payami et al. 1994; Marder et al. 1996; Elbaz et al. 1999). Thus, the development of symptoms in MSA may involve a genetic component.
Homozygous variants (p.M128V-p.V393A/p.M128V-p.V393A) and compound heterozygous variants (p.R387X/p.V393A) in coenzyme Q2, polyprenyltransferase (COQ2) (MIM#609825) were discovered in two families with MSA patients (The Multiple-System Atrophy Research Collaboration 2013). Multiple missense COQ2 variants were also detected in patients with sporadic MSA. COQ2 is located on chromosome 4q21.23 and encodes para-hydroxybenzoate-polyprenyl transferase (Ashby et al. 1992), variants in which induce ubiquinone deficiency (coenzyme Q10 or COQ10) (Quinzii et al. 2006). COQ2 is a catalyzing enzyme in the second step of the reaction sequences in the biosynthesis of COQ10, a fat-soluble substance that functions as an antioxidant, membrane stabilizer, and transporter of electrons from complex I and complex II to complex III in mitochondria. COQ10 deficiency results in various types of neurological disorders such as (1) a myopathic form characterized by myoglobinuria and central nervous system-related symptoms of seizures, ataxia, or mental retardation (Ogasahara et al. 1989; Sobreira et al. 1997), (2) infantile encephalopathy (Rotig et al. 2000), and (3) ataxia and cerebellar atrophy similar to MSA (Musumeci et al. 2001).
The genes associated with familial PD, synuclein alpha (SNCA), parkin, PTEN-induced putative kinase 1 (PINK1), and DJ-1, are strongly related to oxidative stress and mitochondrial dysfunction (Henchcliffe and Beal 2008). In addition, based on the pathological findings, both diseases such as PD and MSA also belong to alpha-synucleinopathies (Savica et al. 2018). Thus, we hypothesized that MSA and PD may share a common pathway that induces neurodegeneration. Herein, we analyzed entire exons and exon–intron boundaries in COQ2 in 123 familial PD, 52 sporadic PD and 39 sporadic MSA cases of Japanese origin. Our results emphasize that COQ2 variants contribute to the pathogenesis of PD.
Methods
Subjects and methods
This study was approved by the ethics review committee of Juntendo University School of Medicine. All participants gave informed and written consent before participation. The study population consisted of 123 patients with familial PD [mean age at onset: 62.8 ± 12.4 years [± standard deviation (SD)], male:female = 57:66], 52 patients with sporadic PD [mean age at onset: 40.6 ± 8.17 years (± SD), male:female = 19:33], and 39 patients with sporadic MSA [mean age at onset: 56.4 ± 10.2 years (± SD), male:female = 16:23]. The cohort of PD cases was further classified as 70 cases with PD with autosomal dominant inheritance [mean age at onset: 59.7 ± 12.8 years (± SD), male:female = 35:35] and 53 with PD with autosomal recessive inheritance [mean age at onset: 66.9 ± 10.6 years (± SD), male:female = 22:31]. All cases were of Japanese origin. The diagnosis of MSA and PD was established on the basis of clinical criteria (Gilman et al. 2008; Hughes et al. 1992). We defined the mode of inheritance as autosomal dominant in cases with affected family members in at least two consecutive generations, and autosomal recessive in cases with affected siblings only in the same generation. None of the enrolled patients had pathogenic variants, multiplication in the entire exon of SNCA, deletions, and multiplications in the entire exons of parkin and variants of PINK1, variants in exon 31, 41, and 48 of leucine-rich repeat kinase 2 (LRRK2), and no risk variants in glucocerebrosidase (GBA).
Sequencing analysis
Genomic DNA was extracted from peripheral blood using QIAamp DNA Blood Maxi Kit (Qiagen, Hilden, Germany). We used primer sets to amplify all coding exons and exon–intron boundaries of COQ2 and sequenced them with the Sanger method using BigDye Terminators v1.1 Cycle Sequencing Kit and 3130 Genetic Analyzer (Life Technologies, Foster City, CA, USA). PCR and sequence primers were designed using Exonprimer (http://ihg.gsf.de/ihg/ExonPrimer.html). The sequences and PCR and reverse-transcriptase PCR conditions are described in supplementary data. The sequences of the subjects were compared with the COQ2 reference sequence (NCBI: NM_015697.7). We evaluated the pathogenicity of each variant using Polyphen-2 (Adzhubei et al. 2013), Mutation Taster (Schwarz et al. 2010), Sorting Tolerant From Intolerant (SIFT) (Li et al. 2009), rare exome variant ensemble learner (REVEL) (Ioannidis et al. 2016), and Combined Annotation Dependent Depletion (CADD) (Kircher et al. 2014). The term ‘pathogenic’ was defined in a previous report (MacArthur et al. 2014). Evolutionary conservation of the mutated amino acids was evaluated using NCBI Homologene (http://www.ncbi.nlm.nih.gov/homologene/). Protein sequence and functional information were evaluated using UniProt (http://www.uniprot.org/).
Association analysis of COQ2 variants for familial cases with MSA and PD
To investigate the contribution of rare non-synonymous COQ2 variants [minor allele frequency (MAF) < 1.0% in public databases] to the pathogenesis of familial PD, we examined and compared the allele frequencies of the COQ2 variants in the familial PD cases versus the data for the east Asian population from Exome Aggregation Consortium (ExAC) (Lek et al. 2016) and that for the Japanese population from Integrated Japanese Genetic Variation Database (IJGVD) (Nagasaki et al. 2015). For statistical analysis, we performed Fisher’s exact test (two-tailed) and calculated the odds ratios and corresponding 95% confidential interval (CI) values to investigate the significant differences between our study samples and public databases. Data were analyzed by SPSS for windows advanced statistics release 6.0 (IBMⓇ, New York, USA).
Results
Variants of COQ2 in PD and MSA
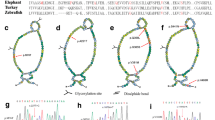
We detected two rare variants in two familial PD cases: Families A and B. Clinical overviews of each patient are summarized in Table 1. The variants were [c.469C > T, p.P157S] of A-IV-1, and [c.43C > A, p.H15N]/[c.991G > A, p.G331S] of B-II-1 (Fig. 1). Three variants were heterozygous. With regard to allele frequencies, p.P157S was not seen in ExAC but was rarely observed in IJGVD (MAF 0.04%), with the prediction of protein damages by amino acid changes underlying the prediction of pathogenicity by in silico analysis (Table 2). For Family B, p.H15N was not seen in ExAC but rarely observed in IJGVD (MAF 0.03%) and did not appear as a pathogenic variant in the REVEL and CADD analysis. The p.G331S variant was recorded as SNPs rs758847245, but the frequency was very rare in the absence of gene database of ExAC and IJGVD. We could not asses the segregation study for Family A and B due to their rejection of our proposal.

Family trees of patients harboring COQ2 variants and the results of direct sequencing of COQ2 in two PD cases. a Family A includes a patient with juvenile Parkinson’s disease (PD) (A-IV-1) with p.P157S. A-III-3 and A-III-6 have consanguinity. A-II-2 was diagnosed with PD. b Family B includes middle-aged onset and mild symptoms of PD with good response to levodopa, with p.H15N/p.G331S. B-I-1 was diagnosed with PD. c C-II-5 was clinically diagnosed as progressive supranuclear palsy-like, harboring exonic skipping of exon 5, p.L261Qfs*4. C-II-1 and C-II-2 was clinically diagnosed with PD. C-II-2 did not have p.L261Qfs*4. d D-II-2 fulfilled criteria of multiple system atrophy with parkinsonism and dysautonomia, harboring p.K407del. Circle with dot or square with dot indicates asymptomatic carriers. The family C and D did not have segregation of COQ2 variants. Three heterozygous missense variants in COQ2, p.P157S (e), p.G331S (f) and p.H15N (g)
We also detected two truncated variants (p.L261Qfs*4 in Family C with PD and p.K407del in Family D with MSA). In Family C, C-II-4 showed a frame shift rearrangement of p.L261Qfs*4, which induced exonic skipping of exon 5, which was confirmed by RT-PCR (supplementary Materials and methods). The clinical manifestation of C-II-4 was late-onset supranuclear palsy-parkinsonism. The clinical diagnosis of C-III-2 was confirmed to be PD with hemi-parkinsonism and a good response to levodopa, but C-III-2 did not show variants of p.L261Qfs*4. The clinical diagnosis for C-II-1 was also PD. Family C did not show segregation of p.L261Qfs*4 and shared different symptoms. In Family D, we clinically diagnosed D-II-2 with MSA-cerebellar type (MSA-C). We collected DNA samples from the patient’s mother (D-I-2), elder sister (D-II-1), and younger brother (D-II-3), who were all asymptomatic carriers. All three shared p.K407del. The details of the four families (Family A-D) are described in the supplementary information. Thus, we concluded that p.L261Qfs*4 and p.K407del did not directly relate to their phenotype. Overall, the allele frequencies of the putative pathogenic variants were 0.8% (2/246) in familial PD and 0% (0/78) in the MSA case in our cohort. Only p.P157S in Family A and p.G331S in Family B seemed to associate with PD.
Association analysis of COQ2 variants between familial PD and MSA versus the public variant database
We identified 11 COQ2 variants as non-synonymous (p.H15N, p.G21S, p. L25V, p.V66L, p.P157S, p.L261Qfs*4, p.G331S, and p.V393A) or synonymous (p.D298D, p.T317T, and p.S330S) for PD, and five variants as non-synonymous (p.V66L, p.V393A, and p.K407del) or synonymous (p.D298D and p.S330S) for MSA (Table 3). Three other variants, namely, p.L261Qfs*4, p.T317T, and p.G331S, were very rare, with no records in the ExAC and IJGVD. The MAFs of four non-synonymous variants, namely, p.H15N, p.G21S, and p.P157S for familial PD and p.K407del for MSA, were under 1.0% in the public variant database. The p.G21S variant showed different frequencies for familial PD versus ExAc (p = 0.004) and versus IJGVD (p = 0.01). After Bonferroni correction, p.G21S showed the most significant association with familial PD. In contrast, p.H15N and p.P157S did not show significance in comparison with IJGVD, but their ORs were both 9.66, and the 95% CI did not cross 1 [p.H15N: 95% CI 1.001–93.159; p.P157S: CI 1.002–93.238]. For MSA patients, p.K407del showed different frequencies in MSA versus ExAc (p = 0.05) (supplementary Table 1). However, our study of Family D indicated that no segregation of p.K407del. p.V393A was frequently observed in sporadic PD (MAF: 7.7%). The p.V393A, which is previously reported risk variant for MSA (The Multiple-System Atrophy Research Collaboration 2013), variant showed different frequencies in all PD (n = 175) versus ExAC (p = 0.07) and versus IJGVD (p = 0.02). The other five variants for PD did not show statistical significance.
Discussion
We detected two rare variants in COQ2, namely p.P157S in Family A and p.G331S in Family B, consisting of patients with juvenile PD and middle-aged onset PD. A-IV-1 with p.P157S manifested as juvenile PD in a woman at 16 years of age, and maintained favorable conditions after 48 years of treatment with levodopa. The patient’s parents were consanguineous. Thus, Family A could carry another variant of the bi-allelic variants of another gene. The p.P157S variant observed in this study was similar to the pathogenic variant reported in MSA (The Multiple-System Atrophy Research Collaboration 2013; Sun et al. 2016). The two mutations p.P157S and p.G331S are very rare in the public gene database and whether they are pathogenic mutations or rare variants still remains a controversy. Among other rare variants, we detected three very rare non-synonymous variants—p.P157S, p.L261Qfs*4, and p.G331S—and one variant significantly seen in familial PD: p.G21S. Our study indicates that COQ2 variants are rarely related to the onset of familial PD. There were no pathogenic variants in patients with MSA.
COQ2 encodes the enzyme in the second step of COQ10 biosynthesis (Quinzii et al. 2006). A missense variant in COQ2 significantly decreases the rate of COQ10 synthesis. COQ2 variant causes primary COQ10 deficiency (Quinzii et al. 2006). These deficiencies in COQ10 are related to various types of neurological disorders, including myopathy, infantile encephalopathy, and ataxia and cerebellar atrophy (Ogasahara et al. 1989; Rotig et al. 2000; Musumeci et al. 2001). These evidences suggest that COQ2 variants affect mitochondrial functions more directly and result in various types of clinical presentations. Indeed, we detected two types of variants from familial PD patients. COQ2 variants may play a crucial role in neurodegenerative disorders via disturbance of redox reactions in the mitochondrial respiratory chain and oxidative stress. Several types of variants have been known to be related to hereditary PD and mitochondrial impairments, such as missense variants or multiplications in SNCA, parkin, PINK1, DJ-1, HtrA serine peptidase 2 (HTRA2), and F-box protein 7 (FBXO7) (Henchcliffe and Beal 2008; Burchell et al. 2013; Conedera et al. 2016). These variants induce instability in mitochondrial maintenance, morphological changes, and decreased levels of dopaminergic neurons via increased oxidative stress. COQ2 variants presumably share a similar pathway and induce the phenotype of PD or parkinsonism.
Regarding the frequency of COQ2 variants in patients with PD, there are insufficient numbers of reports describing the association between the two. Yang et al. (2015) reported a significantly high frequency of p.V393A in patients with PD in China. However, these did not match our data showing a strong association of PD and p.V393V (Table 3). One report described two variants, p.S347C and p.V393A, in 600 PD cases from a European PD cohort (Sharma et al. 2014). There was no association of p.V393 in a comparison of 500 PD cases and 505 age-matched controls in Taiwan (Lin et al. 2015). Sharma et al. (2014) reported p.P157S in one unaffected person in European controls (0.002, 1/600), with no corresponding record in the ExAC gene database. In the comparative study between familial PD and the gene database for the Japanese population, p.G21S is significantly related to familial PD. The frequency seems to be very rare, but p.G21S may contribute to the disease onset as a relative risk factor.
With regard to COQ2 variants in MSA, there were no pathogenic variants in our 39 patients. Just one variant, p.K407del, was seemingly related to MSA patients, but the variant did not show segregation in Family D, clinically presenting symptoms as MSA. Several previous studies could not detect pathogenic COQ2 variants in MSA (Jeon et al. 2014; Sharma et al. 2014; Schottlaender and Houlden 2014). Large population studies also did not support an association between COQ2 variants and MSA (Sailer et al. 2016; Lin et al. 2015), although our analysis support the association. As our speculation, it may be owing to the regional or racial differences of COQ2 variants. Because of the small sample size, our study may not have detected significant differences in variants in MSA. Thus, the association of MSA and COQ2 variants is still debatable. Our study has some limitations. First, the small sample size resulted in statistical weakness. Second, no segregation analysis was conducted for Families A and B. Finally, this was a comparative study that was conducted using data from the public variant database, without age-matched controls.
We reported a genotype–phenotype correlation of COQ2 variants in PD in the Japanese population. We detected two rare variants, p.P157S and p.G331S, in two familial PD cases and a possible risk variant, p.G21S. Our findings may help the understanding of COQ2 variants in familial PD.
Abbreviations
- COQ2 :
-
Coenzyme Q2
- COQ10 :
-
Coenzyme Q10
- PD:
-
Parkinson’s disease
- MSA:
-
Multiple system atrophy
- MRI:
-
Magnetic resonance imaging
References
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 7(Unit7):20. https://doi.org/10.1002/0471142905.hg0720s76
Ashby MN, Kutsunai SY, Ackerman S, Tzagoloff A, Edwards PA (1992) COQ2 is a candidate for the structural gene encoding para-hydroxybenzoate:polyprenyltransferase. J Biol Chem 267(6):4128–4136
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H, Abramov AY, Hardy J, Wood NW, Whitworth AJ, Laman H, Plun-Favreau H (2013) The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16(9):1257–1265. https://doi.org/10.1038/nn.3489
Conedera S, Apaydin H, Li Y, Yoshino H, Ikeda A, Matsushima T, Funayama M, Nishioka K, Hattori N (2016) FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol Aging 40:192.e191–192.e195. https://doi.org/10.1016/j.neurobiolaging.2016.01.003
Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, Dartigues JF, Alperovitch A, Tzourio C, Rocca WA (1999) Familial aggregation of Parkinson’s disease: a population-based case-control study in Europe. EUROPARKINSON Study Group. Neurology 52(9):1876–1882
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372(14):1375–1376. https://doi.org/10.1056/NEJMc1501657
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71(9):670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, Kakita A, Yamada M, Takahashi H, Hirasawa M, Mizuno Y, Ogata K, Goto J, Kanazawa I, Nishizawa M, Tsuji S (2007) Multiplex families with multiple system atrophy. Arch Neurol 64(4):545–551. https://doi.org/10.1001/archneur.64.4.545
Henchcliffe C, Beal MF (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 4(11):600–609. https://doi.org/10.1038/ncpneuro0924
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55(3):181–184
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99(4):877–885. https://doi.org/10.1016/j.ajhg.2016.08.016
Jeon BS, Farrer MJ, Bortnick SF (2014) Mutant COQ2 in multiple-system atrophy. N Engl J Med 371(1):80. https://doi.org/10.1056/NEJMc1311763#SA1
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315. https://doi.org/10.1038/ng.2892
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. https://doi.org/10.1038/nature19057
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics (Oxf, Engl) 25(21):2744–2750. https://doi.org/10.1093/bioinformatics/btp528
MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C (2014) Guidelines for investigating causality of sequence variants in human disease. Nature 508(7497):469–476. https://doi.org/10.1038/nature13127
Marder K, Tang MX, Mejia H, Alfaro B, Cote L, Louis E, Groves J, Mayeux R (1996) Risk of Parkinson’s disease among first-degree relatives: a community-based study. Neurology 47(1):155–160
Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, Weissman BM, Tsao CY, Mendell JR, Shanske S, De Vivo DC, Hirano M, DiMauro S (2001) Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 56(7):849–855
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, Yamaguchi-Kabata Y, Yokozawa J, Danjoh I, Saito S, Sato Y, Mimori T, Tsuda K, Saito R, Pan X, Nishikawa S, Ito S, Kuroki Y, Tanabe O, Fuse N, Kuriyama S, Kiyomoto H, Hozawa A, Minegishi N, Douglas Engel J, Kinoshita K, Kure S, Yaegashi N, Yamamoto M (2015) Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun 6:8018. https://doi.org/10.1038/ncomms9018
Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA 86(7):2379–2382
Payami H, Larsen K, Bernard S, Nutt J (1994) Increased risk of Parkinson’s disease in parents and siblings of patients. Ann Neurol 36(4):659–661. https://doi.org/10.1002/ana.410360417
Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, Hirano M (2006) A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 78(2):345–349. https://doi.org/10.1086/500092
Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P (2000) Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet (Lond Engl) 356(9227):391–395. https://doi.org/10.1016/s0140-6736(00)02531-9
Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, Lees A, Ross OA, Dickson DW, Mok K, Mencacci NE, Schottlaender L, Chelban V, Ling H, O’Sullivan SS, Wood NW, Traynor BJ, Ferrucci L, Federoff HJ, Mhyre TR, Morris HR, Deuschl G, Quinn N, Widner H, Albanese A, Infante J, Bhatia KP, Poewe W, Oertel W, Hoglinger GU, Wullner U, Goldwurm S, Pellecchia MT, Ferreira J, Tolosa E, Bloem BR, Rascol O, Meissner WG, Hardy JA, Revesz T, Holton JL, Gasser T, Wenning GK, Singleton AB, Houlden H (2016) A genome-wide association study in multiple system atrophy. Neurology 87(15):1591–1598. https://doi.org/10.1212/wnl.0000000000003221
Savica R, Bradley BF, Mielke MM (2018) When Do alpha-Synucleinopathies Start? An epidemiological timeline: a review. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2017.4243
Schottlaender LV, Houlden H (2014) Mutant COQ2 in multiple-system atrophy. N Engl J Med 371(1):81. https://doi.org/10.1056/NEJMc1311763#SA3
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576. https://doi.org/10.1038/nmeth0810-575
Sharma M, Wenning G, Kruger R (2014) Mutant COQ2 in multiple-system atrophy. N Engl J Med 371(1):80–81. https://doi.org/10.1056/NEJMc1311763#SA2
Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, Santorelli FM, Miranda AF, Bonilla E, Mojon DS, Barreira AA, King MP, DiMauro S (1997) Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology 48(5):1238–1243
Sun Z, Ohta Y, Yamashita T, Sato K, Takemoto M, Hishikawa N, Abe K (2016) New susceptible variant of COQ2 gene in Japanese patients with sporadic multiple system atrophy. Neurology Genet 2(2):e54. https://doi.org/10.1212/nxg.0000000000000054
The Multiple-System Atrophy Research Collaboration (2013) Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 369(3):233–244. https://doi.org/10.1056/NEJMoa1212115
Vidal JS, Vidailhet M, Derkinderen P, Tzourio C, Alperovitch A (2010) Familial aggregation in atypical Parkinson’s disease: a case control study in multiple system atrophy and progressive supranuclear palsy. J Neurol 257(8):1388–1393. https://doi.org/10.1007/s00415-010-5638-9
Lin CH, Lin HI, Chen ML, Wu RM (2015) COQ2 p. V393A variant, rs148156462, is not associated with Parkinson’s disease in a Taiwanese population. Neurobiol Aging 36(1):546.e517–546.e548. https://doi.org/10.1016/j.neurobiolaging.2014.08.006
Acknowledgements
KK is funded by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (23591269, 26461319). This work was supported by JSPS KAKENHI Grant numbers, 16K09678 (to KN), 16K09700 (to YL), 16K09676 (to MF), and 15H04842 (to NH). We are very grateful for these Grants: AMED-CREST (Japanese Association of Medical Research and Development) (N.H.), Practical Research Project for Rare/Intractable Diseases from AMED; 15ek0109029s0202 to NH.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors report no conflicts of interest relevant to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mikasa, M., Kanai, K., Li, Y. et al. COQ2 variants in Parkinson’s disease and multiple system atrophy. J Neural Transm 125, 937–944 (2018). https://doi.org/10.1007/s00702-018-1885-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-018-1885-1