
Abstract
Parkinson’s disease (PD) is a devastating disorder, affecting approximately 2% of people aged 60 and above. It is marked by progressive neurodegeneration that has long been known to impact dopaminergic cells and circuits, but more recently the acetylcholine system has also been implicated in the complex aetiology and symptomatology of the disease. While broad changes in cholinergic markers have been described, insight into the contribution of specific acetylcholine receptors is less clear. To address this important unknown, in this study we performed [3H] pirenzepine, [3H] 4DAMP, and [3H] AF-DX 384 in situ radioligand binding on postmortem tissues from Brodmann’s area 6, 9, 46, and the caudate putamen, from PD and matched controls to detect muscarinic M1, M3, and M1/2/4 receptors, respectively. We found no difference in [3H] pirenzepine binding between PD and controls across all regions assessed. [3H] 4DAMP binding was found to be higher in PD CPu and BA9 than in controls. [3H] AF-DX 384 was higher in BA9 of PD compared with controls. In sum, we show selective increase in M3 receptors in cortical and subcortical regions, as well as increased M2/M4 in cortical area BA9, which together support a role for cholinergic dysfunction in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive degenerative disorder of the central nervous system (CNS), characterized by motor, psychiatric, and cognitive impairment. While an impaired dopaminergic system explains some aspects of the symptomatology, the pathophysiology of the full spectrum is not yet delineated and therefore, there is increasing interest in the role of other neurotransmitter systems in the genesis of the symptoms of the disorder. Both cognition and motor perseveration are affected in early PD, and it is notable that both are modulated by the cholinergic system (Stoffers et al. 2001). Hence, there have been efforts to identify changes in markers of the cholinergic system in PD as a step toward understanding how cholinergic function may be affected.
Postmortem studies in the CNS from subjects with PD have investigated the presynaptic marker choline acetyltransferase (ChAT) as well as the two families of receptors that are targeted by acetylcholine; the nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs). The nAChRs are ligand gated ion channels found throughout the brain in homomeric or heteromeric conformations. Studies have reported lower levels of nAChRs in cortex, hippocampus, substantia nigra, and striatum of PD subjects (Aubert et al. 1992; Perry et al. 1990; Pimlott et al. 2004). These decreases have been attributed to a loss of neuronal integrity and are most pronounced for α6β2* nAChRs (reviewed in Quik et al. 2011). The other family of cholinergic receptors is the muscarinic acetylcholine receptors (mAChRs), made up of five G protein-coupled receptors (M1–M5). Significantly, neuroimaging studies using a non-selective radioligands have reported higher mAChRs in frontal cortex (Asahina et al. 1998) and occipital lobe (Colloby et al. 2006) in PD. These data are supported by some studies in postmortem CNS (Lange et al. 1989, 1993; Perry et al. 1990; Ruberg et al. 1982). However, the contribution of the specific mAChRs subtypes is less clear, as relatively few investigations have attempted to determine whether all mAChR are differentially expressed in PD, or whether select subtypes underlie these changes (Aubert et al. 1992; Joyce 1993; Perry et al. 1990; Rinne et al. 1989). Importantly, muscarinic antagonists can be clinically effective in PD (Fahn et al. 1990), highlighting the relevance of further investigations into this receptor (Dencker et al. 2012).
We have carried out extensive studies on muscarinic receptors in the CNS from subjects with schizophrenia and have used varying assay conditions and tissue from muscarinic receptor knock-out mice to increase the specificity of binding for commonly used muscarinic receptor ligands. Thus, in the assays we use we have been able to increase the specificity of [3H]pirenzepine binding to the muscarinic M1 receptor in human CNS tissue to ~80% (Gibbons et al. 2013; Scarr and Dean 2008) (Crook et al. 2001) and to make [3H]4DAMP binding very selective for the muscarinic M3 receptor (Jeon et al. 2013). In the present study, we used these methodologies along with a non-modified [3H]AF-DX protocol to determine if there are subtype specific changes in levels of M1, M2/M4, and M3 subtypes of mAChRs in brain regions associated with PD pathology and symptomatology.
Methods
Human tissue collection
Following consent from the Ethics Committee of the Victorian Institute of Forensic Medicine and the Mental Health Research and Ethics Committee of Melbourne Health, postmortem samples were obtained from the Victorian Brain Bank Network (VBBN). The left hemispheres of human CNS were collected postmortem, rapidly sliced, and frozen to −80 °C as described previously (Dean et al. 1999).
All patients came to the clinic with a diagnosis of PD. Postmortem neuropathological diagnosis of PD was made using the standard criteria of neuronal loss, pigment incontinence, and the presence of α-synuclein reactive Lewy Bodies in the substania nigra. All patients came to VBBN with a clinical diagnosis of PD. None of the subjects were diagnosed with associated Alzheimer’s disease, but most had scattered Amyloid beta plaques, as is commonly observed with aging. 8 of the 11 cases also presented with Lewy bodies in the cortex. These were limited in number, and the clinical significance is difficult to predict.
Brodmann’s Area 6 (BA6), BA9, BA46, and Caudate Putamen (CPu) were selected for study based upon their known involvement in PD, their anatomy, or involvement in perseverative, cognitive behavior.
BA6 was defined as the caudal superior frontal gyrus and the middle frontal gyrus extending from the cingulate sulcus on the medial surface to the lateral sulcus on the lateral surface. BA9 (bridging the dorsolateral prefrontal cortex and the medial prefrontal cortex) was bounded by the lateral surface of the frontal lobe, comprising the middle frontal gyrus superior to the inferior frontal sulcus. Dorsolateral prefrontal cortical BA46 was defined as within the lateral surface of the frontal lobe approximately constrained to the middle third of the middle frontal gyrus and the most rostral inferior frontal gyrus. Caudate putamen is a distinct region.
Cadavers were stored at 4 °C within 5 h of death. During case history, review age at death, gender, post mortem interval (PMI), and brain pH (Kingsbury et al. 1995) were determined (see Table 1). With regard to PMI, where death was witnessed, PMI was from time of death to autopsy, but if the death was not witnessed, time of death was taken as the mid-point between the time the donor was last observed alive (maximum of 5 h was allowed), and the time found dead. Cohorts were matched as closely as possible for age, PMI, and brain pH.
Autoradiographic ligand binding
Autoradiography was performed upon 20 µm sections cut using a Leica cryostat onto gelatinised slides. Frozen sections were stored at −70 °C. On the day of the experiment, sections were removed from −70 °C and air-dried at room temperature for 1 h prior to radioligand specific protocols (described below). Following the final wash, slides were dried under a stream of cool air, and fixed overnight in a desiccator containing paraformaldehyde powder. Slides were then apposed to phosphorimaging plates. Microscales were used to provide a frame of reference for measuring radioactivity, and allowing us to normalize values across plates and across experiments. After exposure, plates were scanned with a BAS5000, and density was established using AIS software.
[3H]pirenzepine autoradiography
The methodology use for [3H]pirenzepine binding was modified to increase selectivity for the muscarinic M1 receptor to ~80% (Gibbons et al. 2013; Scarr and Dean 2008); thus, frozen tissue sections were incubated in 15 nM [3H]Pirenzepine ([3H]PZP) in assay buffer (10 mM KH2PO4, 10 mM Na2PO4, pH 7.4) in the absence [total binding (TB)] or presence [non-specific binding (NSB)] of 1 mM 3-quinuclidinyl xanthene-9-carboxylate hemioxalate hydrate (QNX) (a broad spectrum muscarinic receptor binding ligand to block non-specific binding). Following this incubation, sections were washed twice for 2 min in ice-cold assay buffer, dipped in ice-cold ddH2O. Slides were apposed to phosphorimaging plates with microscales for 3 days.
[3H]4DAMP
The [3H]-DAMP methodology used in this study was developed to increase selectivity of the radioligand for the muscarinic M3 receptor (Jeon et al. 2013). Hence, sections were preincubated for 15 min in assay buffer (50 mM Tris–HCl, pH7.4), rinsed in ddH2O, and air-dried at room temperature for 1 h. Subsequently, sections were incubated in assay buffer containing 6 nM [3H]4DAMP + 1uM PZP (to block non-specific binding to M1/M4) in the absence (TB) or presence (NSB) of 10 µM 4DAMP mustard for 60 min at room temperature. Slides were then washed twice for 5 min in ice-cold assay buffer, and dipped in ice-cold ddH2O for 15 s. Slides were apposed to phosphorimaging plates for 3 days.
[3H]AF-DX 384
The [3H]AF-DX 384 protocol used in this study detects M1, M2, and M4 receptors. Sections were preincubated for 30 min in Assay Buffer (10 mM KH2PO4, 10 mM Na2PO4, pH 7.4) at room temperature, rinsed in ddH2O and air-dried. Sections were incubated in assay buffer containing 7 nM [3H]AF-DX 384 in the absence (TB) or presence of 1 µM tropicamide (NSB) for 60 min at room temperature, washed twice for 2 min each in ice-cold assay buffer, and dipped in ice-cold water (15 s). Slides were apposed to plates for 7 days.
Statistical analyses
Data were analysed using the D’Agostino Pearson normality test, to determine Gaussian distribution. Pearson’s correlation analysis was performed to determine whether binding in different brain regions was independent, and to identify covariates (PMI, age, and pH). An R 2 value greater than 0.49 was considered a strong relationship (Cook and Weisberg 1999). T tests were used to determine whether gender had a significant association with binding levels. A P value of <0.05 was considered significant.
Significant correlations were shown between regions with each ligand. Given that regions were not independent variables, T tests were thus performed for each region, to determine the influence of diagnosis over binding. If a group failed the normality test, diagnostic differences were analysed by Mann–Whitney test. Where an overall effect was detected, Cohen’s d effect size was calculated. Data are represented as mean ± SEM. With the exception of Cohen’s d, all statistical analyses were performed using Prism 6.0. Cohen’s d was calculated using an online tool available at http://www.cognitiveflexibility.org, and ANCOVA was performed in minitab.
Results
Cohort demographics
The cohort comprised of ten control subjects (five male and five female) and ten subjects who had a diagnosis of PD (five male and five female) (Tables 1, 2). Age at death did not differ according to diagnosis, or did PMI. Variance also did not differ according to diagnosis (Age F = 1.181, P = 0.8087, PMI F = 1.298, P = 0.7042).
pH was significantly higher (P < 0.01, t = 3.494, df = 18) in the PD cohort (6.52 ± 0.06) than in controls (6.22 ± 0.06). Variance did not differ between groups (P = 0.76, F = 1.23).
[3H]PZP
In BA6, [3H]PZP binding did not vary between controls and PD (t = 0.7085, df = 18, P = 0.4428)(Fig. 1a); F tests to compare variance showed no significant difference (F = 1.009, DFn = 9, Dfd = 9, P = 0.99). In BA9, [3H]PZP binding was similar in PD and controls (Fig. 1b, Mann–Whitney test: P = 0.28). In BA46, [3H]PZP binding was similar in PD and controls (Fig. 1c, t = 1.062, df = 18, P = 0.30). Variance also did not differ between diagnostic groups (F = 1.559, DFn = 9, Dfd = 9, P = 0.52). [3H]PZP binding in the CPu was similar in PD and controls (Fig. 1d, Mann–Whitney: P = 0.89) (Table 3).

[3H]Pirenzepine binding in Parkinson’s disease and controls. [3H]Pirenzepine (PZP) binding was calculated by subtracting non-specific binding (inset images) from total binding. [3H]PZP binding was not different in Parkinson’s disease compared with controls in BA 6 (a), BA 9 (b), BA 46 (c), or CPu (d)
Sex, age, pH, or PMI and did not influence [3H]PZP binding in any of the regions assessed (Table 4).
[3H]4DAMP
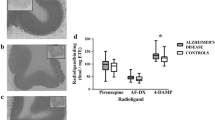
[3H]4DAMP binding did not differ by diagnosis in BA6 (t = 1.67, df = 18, P = 0.11 (Fig. 2a). F tests to compare variance showed no significant difference (F = 1.02, DFn = 9, Dfd = 9, P = 0.97). In BA9, [3H]4DAMP binding was significantly higher in PD than in controls (Fig. 2b, t = 3.37, df = 18, P < 0.01, Cohen’s d = −4.77). Variances did not differ between groups (F = 1.06, DFn = 9, Dfd = 9, P = 0.93). In BA46, [3H]4DAMP binding was similar in controls and PD subjects (Fig. 2c, t = 1.35, df = 18, P = 0.19). Variance did not differ between diagnostic groups (F = 1.55, DFn = 9, Dfd = 9, P = 0.53). In the CPu (Fig. 2d), [3H]4DAMP binding was significant higher in PD compared to controls (t = 2.30, df = 18, P < 0.05, Cohen’s d = −3.28). No difference in variance was detected (F = 1.71, Dfn = 9, Dfd = 9, P = 0.44) (Table 3).

[3H]4DAMP binding in Parkinson’s disease and controls. [3H]4DAMP binding was calculated by subtracting non-specific binding (inset images) from total binding. Binding was significantly higher in Parkinson’s disease (PD) group in BA 9 relative to control (b, P < 0.01). Higher binding was also detected in the caudate putamen (d, P < 0.05) of PD, compared to controls. No changes were observed in BA 6 (a), or in BA 46 (c)
There were no strong correlations between [3H]4DAMP binding and sex, age, pH, or PMI in any region (Table 4).
[3H]AF-DX 384
BA6 [3H]AF-DX 384 binding was similar in PD and controls (Fig. 3a, t = 1.90, df = 18, P = 0.07). F tests to compare variance showed no significant difference (F = 1.09, DFn = 9, Dfd = 9, P = 0.89). In BA9, AF-DX 384 binding levels were significantly higher in the PD diagnostic group than in control subjects (Fig. 3b, t = 2.60, df = 18, P = 0.02, Cohen’s d = −3.67). Variances did not differ between groups (F = 1.06, DFn = 9, Dfd = 9, P = 0.93). In BA46, no significant differences were detected for AF-DX 384 binding (Fig. 3c, t = 0.97, df = 18, P = 0.34). Variance did not differ between diagnostic groups (F = 1.90, DFn = 9, Dfd = 9, P = 0.38). No difference in AF-DX 384 binding was observed in the CPu (Fig. 3d), and no difference in variance was detected (F = 1.43, Dfn = 9, Dfd = 9, P = 0.60) (Table 3).

[3H]AF-DX 384 binding in Parkinson’s disease and controls. [3H]AF-DX 384 binding was calculated by subtracting non-specific binding (inset images) from total binding. We observed significantly higher [3H]AF-DX 384 binding in BA 9 in Parkinson’s disease compared to controls (b, P < 0.05). No differences were observed in BA 6 (a), BA 46 (c), or the caudate putamen (CPu) (d)
Again, there were no strong correlations between sex, age, and PMI and [3H]AF-DX 384 binding in any CNS region. There was a significant relationship between AF-DX 384 binding and pH in BA9 (Table 4). ANCOVA was performed and significance by diagnosis was lost but still was a strong trend (P = 0.054).
Discussion
Dysregulation of the cholinergic system is emerging as a potential pathophysiological mechanism in PD. In this study, we showed higher levels of [3H]4-DAMP and [3H]AF-DX 384 binding in BA9 and higher [3H]4-DAMP binding in CPu. We have shown that [3H]4DAMP binding under the conditions used in our study is selective for the muscarinic M3 receptor (Jeon et al. 2013). We have also shown that under the conditions used this study [3H]AF-DX 384 would bind to the muscarinic M1 as well as the M2 and M4 receptors (Gibbons et al. 2013). Thus, together with the absence of change in [3H]PZP binding to M1, these findings suggest higher M2, M3 and/or M4 in BA9, and higher M3 in CPu.
This conclusion can be further refined, with reference to the literature. Decreased ChAT has been reported in PD postmortem tissue in many regions (Hall et al. 2014; Mufson et al. 1991; Perry et al. 1986; Rinne et al. 1989) suggesting a loss of cholinergic neurons, accompanied by a loss of presynaptic muscarinic receptors. M2 and M4 receptors are both considered to localize presynaptically, acting as autoreceptors; however, M2 appears to predominate in this role in the cortex, while M4 does so in the CPu (Zhang et al. 2002). Our finding of increased [3H]AF-DX 384 binding in BA9 may indicate an increase in M2, M4, or M2 and M4 in an attempt to compensate for lower levels of ACh linked to lower levels of innervating cholinergic neurons, as suggested by decreased ChAT.
Considerable evidence for changes in cholinergic function in PD has been generated in patients both with and without dementia; however, a consistent picture is yet to emerge (Asahina et al. 1998; Colloby et al. 2006; Hilker et al. 2005; Kuhl et al. 1996; Lange et al. 1989, 1993; Perry et al. 1990; Ruberg et al. 1982; Shinotoh et al. 1999). Aligning with our findings, studies using radioligand binding to tissue homogenates found M1/3/5 and M2/4 receptors to be increased in various brain regions, accompanied by a decrease in ChAT activity, and that these changes may depend upon the cognitive state of the patient (Perry et al. 1986) (Rinne et al. 1989). [3H]PZP (Aubert et al. 1992) binding in cortical and subcortical regions has been shown to be similar in PD compared with controls (Aubert et al. 1992). In contrast, Aubert et al. (1992) showed decreased [3H]AF-DX 384 binding in the cortex of PD (but comparable levels in the striatum). Studies performed using in situ radioligand binding, have reported unchanged [3H]AF-DX 384 binding in cortical and subcortical regions in PD compared with controls (Piggott et al. 2003), as well as lower [3H]PZP and [3H]NMS binding in the CPu (Joyce 1993).
These divergent findings could result from methodological differences between studies. Aubert et al. performed saturation binding analysis on alternating hemispheres of BA9 and BA10 tissue homogenate to determine receptor level. We did not study BA10 and it is possible that mAChR density differs in the two hemispheres, which might explain the findings. Importantly, in the present study, the protocol for [3H]PZP binding has been optimized for the detection of M1 receptors over other mAChRs and possesses several methodological differences from the older investigation that used fewer subjects, a lower concentration of [3H]PZP, and an unspecified incubation time. Finally, the stage of PD and the presence or absence of dementia and/or neuropsychiatric symptoms at the time of death could very well affect mAChR expression; it has been proposed that increases in mAChRs are a compensatory mechanism that decelerates the development of cognitive symptoms. In this way, the changes described above may capture different symptomatological groups of patients; patients with cognitive impairment may show no changes, or decreases in mAChRs while increases may protect against the development of this symptom subset, at least transiently. Clinically, cognitive impairment has been proposed to arise from impaired ability to maintain a cognitive set, difficulty in switching tasks, and a tendency to perseverate (Alevriadou et al. 1999; Cools et al. 1984; Ebersbach et al. 1994); however, in the present study, the lack of data on cognitive performance of these patients prevents correlation between cognition and neurochemistry.
Interestingly, we found the levels of binding to M2, M3 and M4 receptors to be altered, whereas binding to M1, a receptor heavily implicated in cognition, was not. However, the importance of other cholinergic receptors in cognition should not be ignored. From a neuroanatomical perspective, in the present study, the greatest cumulative difference was observed in BA9, which showed higher levels of M2/M4 and M3 receptors, compared with controls. Consistent with a role for mAChR in the symptomatology of PD, BA9 has been implicated as having a role in many domains of cognition (MacDonald et al. 2000). The emphasis on the association between cholinergic denervation and cognition should not obscure the relationships between the cholinergic system and olfaction, sleep, affect, and motor function all of which are affected in PD. Our finding of increased M3 in the CPu may speak to the motor symptoms that develop as part of Parkinson’s disease course, as this region has been convincingly shown to play a key role in this dysfunction (Shadmehr and Krakauer 2008).
Studies in knock-out mouse lines for all five mAChR paint a picture of intricate, regulated control over brain function and behavior. Both M2 and M4 KO mice show mitigation of pharmacologically induced tremulous jaw movements, an endophenotype of parkinsonian tremor (Bymaster et al. 2001; Salamone et al. 2001). M2 receptors have also been implicated in cognition; M2 KO mice demonstrate significant deficits in behavioral flexibility and impairment in passive avoidance tasks (Tzavara et al. 2003). In contrast, M2 antagonism improved learning performances in both rats with scopolamine-induced amnesia and in aged memory-impaired rats (Doods et al. 1993; Quirion et al. 1995), highlighting the potential for compensatory changes to buffer imbalances in the cholinergic system. Behavioral studies have also implicated M3 in maintaining cognitive function (Fedoce et al. 2016; Poulin et al. 2010) however, little else is known about the role of central M3 receptors. Hence, if animal studies can be extrapolated to the human condition, the higher levels of M2 and/or M4 receptors in BA 9 might be involved in tremor and cognition, whereas changes in M3 are more focused on cognition.
We must also consider the relationship between PD, [3H]AF-DX 384 binding, and pH observed in BA9. ANCOVA analyses suggest that the significance detected in this region may be, at least partly, associated with differences in pH across diagnosis; further experiments will be required to determine if changes in [3H]AF-DX 384 binding relate to diagnosis or CNS pH.
The M3 is the least well characterized of the mAChRs and the present study, showing significant increases in [3H]4-DAMP binding, is the only study that we are aware of describing altered expression of this receptor in PD. The changes in mAChR described in the present study are also likely to directly impact dopaminergic activity, exacerbating disruptions of dopaminergic tone in these regions (Zhang et al. 2002), further aggravating the symptomatology, and underscoring the interconnectedness of these two systems.
The classical hypothesis of acetylcholine’s contribution to the pathophysiology of PD posits that change in acetylcholine is secondary to changes in dopamine; decreased dopaminergic tone in the striatum releases inhibition over cholinergic neurons, resulting in cholinergic increases (Emre et al. 2004). More recently, a ‘dual syndrome hypothesis’ proposed that two distinct streams of dysfunction arise from cholinergic or dopaminergic deficits, contributing to different subsets of symptoms. This latter theory is rapidly gaining ground, as research establishes AChR deficits early in the disorder, distinct from DA dysregulation; however, the relationship between dopaminergic and cholinergic deficits is likely to be much more complicated. Here, we show increased [3H]AF-DX 384 binding to M2/M4 in BA9, and increased [3H]4DAMP binding to M3 in BA9 and CPu, with no changes in [3H]PZP. These findings support an important role for muscarinic acetylcholine receptors in the pathophysiology of PD, underscoring the need for further investigations into their contribution to disease development and progression.
References
Alevriadou A, Katsarou Z, Bostantjopoulou S, Kiosseoglou G, Mentenopoulos G (1999) Wisconsin Card Sorting Test variables in relation to motor symptoms in Parkinson’s disease. Percept Mot Skills 89(3 Pt 1):824–830. doi:10.2466/pms.1999.89.3.824
Asahina M, Suhara T, Shinotoh H, Inoue O, Suzuki K, Hattori T (1998) Brain muscarinic receptors in progressive supranuclear palsy and Parkinson’s disease: a positron emission tomographic study. J Neurol Neurosurg Psychiatry 65(2):155–163
Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R (1992) Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem 58(2):529–541
Bymaster FP, Carter PA, Zhang L, Falcone JF, Stengel PW, Cohen ML, Shannon HE, Gomeza J, Wess J, Felder CC (2001) Investigations into the physiological role of muscarinic M2 and M4 muscarinic and M4 receptor subtypes using receptor knockout mice. Life Sci 68(22–23):2473–2479
Colloby SJ, Pakrasi S, Firbank MJ, Perry EK, Piggott MA, Owens J, Wyper DJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT (2006) In vivo SPECT imaging of muscarinic acetylcholine receptors using (R, R) 123I-QNB in dementia with Lewy bodies and Parkinson’s disease dementia. NeuroImage 33(2):423–429. doi:10.1016/j.neuroimage.2006.07.026
Cook RD, Weisberg S (1999) Applied regression including computing and graphics. Wiley series in probability and statistics texts and references section. Wiley, New York
Cools AR, van den Bercken JH, Horstink MW, van Spaendonck KP, Berger HJ (1984) Cognitive and motor shifting aptitude disorder in Parkinson’s disease. J Neurol Neurosurg Psychiatry 47(5):443–453
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2001) Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of Brodmann’s areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am J Psychiatry 158(6):918–925. doi:10.1176/appi.ajp.158.6.918
Dean B, Pavey G, Chai SY, Mendelson FAO (1999) The localisation and quantification of molecular changes in the human brain using in situ radioligand binding and autoradiography. In: Dean B, Kleinman JE, Hyde TM (eds) Using CNS tissue in psychiatric research. Harwood Academic Press, Sydney, pp 67–83
Dencker D, Thomsen M, Wortwein G, Weikop P, Cui Y, Jeon J, Wess J, Fink-Jensen A (2012) Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse and Parkinson’s disease. ACS Chem Neurosci 3(2):80–89. doi:10.1021/cn200110q
Doods H, Entzeroth M, Ziegler H, Schiavi G, Engel W, Mihm G, Rudolf K, Eberlein W (1993) Characterization of BIBN 99: a lipophilic and selective muscarinic M2 receptor antagonist. Eur J Pharmacol 242(1):23–30
Ebersbach G, Hattig H, Schelosky L, Wissel J, Poewe W (1994) Perseverative motor behaviour in Parkinson’s disease. Neuropsychologia 32(7):799–804
Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP, Durif F, Kulisevsky J, van Laar T, Lees A, Poewe W, Robillard A, Rosa MM, Wolters E, Quarg P, Tekin S, Lane R (2004) Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 351(24):2509–2518. doi:10.1056/NEJMoa041470
Fahn S, Burke R, Stern Y (1990) Antimuscarinic drugs in the treatment of movement disorders. Prog Brain Res 84:389–397
Fedoce AG, Ferreira-Junior NC, Reis DG, Correa FM, Resstel LB (2016) M3 muscarinic receptor in the ventral medial prefrontal cortex modulating the expression of contextual fear conditioning in rats. Psychopharmacology 233(2):267–280. doi:10.1007/s00213-015-4109-5
Gibbons AS, Scarr E, Boer S, Money T, Jeon WJ, Felder C, Dean B (2013) Widespread decreases in cortical muscarinic receptors in a subset of people with schizophrenia. Int J Neuropsychopharmacol 16(1):37–46. doi:10.1017/S1461145712000028
Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D (2014) Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain J Neurol 137(Pt 9):2493–2508. doi:10.1093/brain/awu193
Hilker R, Portman AT, Voges J, Staal MJ, Burghaus L, van Laar T, Koulousakis A, Maguire RP, Pruim J, de Jong BM, Herholz K, Sturm V, Heiss WD, Leenders KL (2005) Disease progression continues in patients with advanced Parkinson’s disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 76(9):1217–1221. doi:10.1136/jnnp.2004.057893
Jeon WJ, Gibbons AS, Dean B (2013) The use of a modified [3H]4-DAMP radioligand binding assay with increased selectivity for muscarinic M3 receptor shows that cortical CHRM3 levels are not altered in mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 47:7–12. doi:10.1016/j.pnpbp.2013.08.001
Joyce JN (1993) Differential response of striatal dopamine and muscarinic cholinergic receptor subtypes to the loss of dopamine. III. Results in Parkinson’s disease cases. Brain Res 600(1):156–160
Kingsbury AE, Foster OJ, Nisbet AP, Cairns N, Bray L, Eve DJ, Lees AJ, Marsden CD (1995) Tissue pH as an indicator of mRNA preservation in human post-mortem brain. Brain Res Mol Brain Res 28(2):311–318
Kuhl DE, Minoshima S, Fessler JA, Frey KA, Foster NL, Ficaro EP, Wieland DM, Koeppe RA (1996) In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann Neurol 40(3):399–410. doi:10.1002/ana.410400309
Lange KW, Wells FR, Rossor MN, Jenner P, Marsden CD (1989) Cortical muscarinic receptors in demented patients with Alzheimer’s disease or Parkinson’s disease. Br J Pharmacol 98(Suppl):817P
Lange KW, Wells FR, Jenner P, Marsden CD (1993) Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson’s disease. J Neurochem 60(1):197–203
MacDonald AW 3rd, Cohen JD, Stenger VA, Carter CS (2000) Dissociating the role of the dorsolateral prefrontal and anterior cingulate cortex in cognitive control. Science 288(5472):1835–1838
Mufson EJ, Presley LN, Kordower JH (1991) Nerve growth factor receptor immunoreactivity within the nucleus basalis (Ch4) in Parkinson’s disease: reduced cell numbers and co-localization with cholinergic neurons. Brain Res 539(1):19–30
Perry EK, Perry RH, Smith CJ, Purohit D, Bonham J, Dick DJ, Candy JM, Edwardson JA, Fairbairn A (1986) Cholinergic receptors in cognitive disorders. Can J Neurol Sci 13(4 Suppl):521–527
Perry EK, Smith CJ, Court JA, Perry RH (1990) Cholinergic nicotinic and muscarinic receptors in dementia of Alzheimer, Parkinson and Lewy body types. J Neural Transm Park Dis Dement Sect 2(3):149–158
Piggott MA, Owens J, O’Brien J, Colloby S, Fenwick J, Wyper D, Jaros E, Johnson M, Perry RH, Perry EK (2003) Muscarinic receptors in basal ganglia in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease. J Chem Neuroanat 25(3):161–173
Pimlott SL, Piggott M, Owens J, Greally E, Court JA, Jaros E, Perry RH, Perry EK, Wyper D (2004) Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[(125)i]-a-85380. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 29(1):108–116. doi:10.1038/sj.npp.1300302
Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R, Kong KC, Bottrill A, Mistry S, Wess J, Rosethorne EM, Charlton SJ, Tobin AB (2010) The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc Natl Acad Sci USA 107(20):9440–9445. doi:10.1073/pnas.0914801107
Quik M, Bordia T, Huang L, Perez X (2011) Targeting nicotinic receptors for Parkinson’s disease therapy. CNS Neurol Disord Drug Targets 10(6):651–658
Quirion R, Wilson A, Rowe W, Aubert I, Richard J, Doods H, Parent A, White N, Meaney MJ (1995) Facilitation of acetylcholine release and cognitive performance by an M(2)-muscarinic receptor antagonist in aged memory-impaired. J Neurosci 15(2):1455–1462
Rinne JO, Lonnberg P, Marjamaki P, Rinne UK (1989) Brain muscarinic receptor subtypes are differently affected in Alzheimer’s disease and Parkinson’s disease. Brain Res 483(2):402–406
Ruberg M, Ploska A, Javoy-Agid F, Agid Y (1982) Muscarinic binding and choline acetyltransferase activity in Parkinsonian subjects with reference to dementia. Brain Res 232(1):129–139
Salamone JD, Correa M, Carlson BB, Wisniecki A, Mayorga AJ, Nisenbaum E, Nisenbaum L, Felder C (2001) Neostriatal muscarinic receptor subtypes involved in the generation of tremulous jaw movements in rodents implications for cholinergic involvement in parkinsonism. Life Sci 68(22–23):2579–2584
Scarr E, Dean B (2008) Muscarinic receptors: do they have a role in the pathology and treatment of schizophrenia? J Neurochem 107(5):1188–1195. doi:10.1111/j.1471-4159.2008.05711.x
Shadmehr R, Krakauer JW (2008) A computational neuroanatomy for motor control. Exp Brain Res 185(3):359–381. doi:10.1007/s00221-008-1280-5
Shinotoh H, Namba H, Yamaguchi M, Fukushi K, Nagatsuka S, Iyo M, Asahina M, Hattori T, Tanada S, Irie T (1999) Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson’s disease and progressive supranuclear palsy. Ann Neurol 46(1):62–69
Stoffers D, Berendse HW, Deijen JB, Wolters EC (2001) Motor perseveration is an early sign of Parkinson’s disease. Neurology 57(11):2111–2113
Tzavara ET, Bymaster FP, Felder CC, Wade M, Gomeza J, Wess J, McKinzie DL, Nomikos GG (2003) Dysregulated hippocampal acetylcholine neurotransmission and impaired cognition in M2, M4 and M2/M4 muscarinic receptor knockout mice. Mol Psychiatry 8(7):673–679. doi:10.1038/sj.mp.4001270
Zhang W, Yamada M, Gomeza J, Basile AS, Wess J (2002) Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1–M5 muscarinic receptor knock-out mice. J Neurosci 22(15):6347–6352 (doi:20026644)
Acknowledgements
We would like to thank the families of the subjects for donating the tissue used in these studies. Human brain tissues were received from the Victorian Brain Bank Network, supported by The Florey Institute of Neuroscience and Mental Health, The Alfred and the Victorian Forensic Institute of Medicine, and funded by Australia’s National Health & Medical Research Council and Parkinson’s Victoria. We would also like to thank Andrew Gibbons, Natalie Thomas, and Won Je Jeon for their input on the radioligand binding methodology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
CEM was supported by an NHMRC overseas biomedical research fellowship (APP628906), a Brain and Behavior Research Foundation Young Investigator Award (19543), while these studies were being performed. BD is an NHMRC Senior Research Fellow (APP1002240). This work was supported in part by NHMRC Project Grants (APP1045619, APP628699, and APP1066144), the Victorian Government’s Operational Infrastructure Support, and the Rebecca Cooper Medical Research Foundation. ES was supported by an ARC future fellowship (FT100100689). The funding sources were not involved in the study design; in the collection, the analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed, and all procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Rights and permissions
About this article

Cite this article
McOmish, C., Pavey, G., McLean, C. et al. Muscarinic receptor binding changes in postmortem Parkinson’s disease. J Neural Transm 124, 227–236 (2017). https://doi.org/10.1007/s00702-016-1629-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-016-1629-z