
Abstract
Background
Supra-tentorial primitive neuroectodermal tumors (SPNET) are high-grade, hemispheric tumors, which account for around 2–3 % of pediatric brain tumors. We herein intend to report the clinical features and treatment outcome of patients with nonpineal SPNET treated at our institute.
Methods
Clinical data were collected by retrospective chart review from 2006 to 2012. Histopathology slides were reviewed, and relevant immunohistochemistry stains were done. Overall survival (OS), recurrence-free survival (RFS) and event-free survival (EFS) were analyzed by the Kaplan-Meier product-limit method.
Results
Fifteen patients met the study criterion (male: female = 2:1). Median age at presentation was 11 years (range 3–49 years). Surgical resection was gross total in 6 (40 %) and subtotal in 8 (53.33 %) patients. At presentation, two patients had leptomeningeal dissemination. Radiation therapy was delivered in 11 (73.33 %) patients: craniospinal irradiation in 8 (36 Gy/20 fractions/4 weeks to the craniospinal axis followed by a local boost of 20 Gy/10 fractions/2 weeks) and focal RT in 3 patients. Systemic chemotherapy (median 6 cycles; range 1–16 cycles), given in 13 (86.67 %) patients, included the VAC regimen (vincristine, adriamycin, cyclophosphamide) alternating with IE (ifosfamide,etoposide). After a median follow-up of 22.6 months (mean, 24.47 months), complete response and progressive disease were noted in 8 (53.33 %) and 7 (46.67 %) patients, respectively. Median OS was not reached, and estimated median EFS was noted to be 4.12 years (actuarial rate of EFS at 2 years, 55.2 %).
Conclusion
Maximal safe resection followed by craniospinal irradiation and systemic chemotherapy with 6–12 cycles of an alternating regimen of VAC and IE is a reasonable treatment strategy in patients with nonpineal SPNET.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Supratentorial primitive neuroectodermal tumor (SPNET) is a high-grade central nervous system malignancy that makes up approximately 2–3 % of all childhood brain tumors [11]. It was first described as a separate disease entity by Hart and Earle in 1973 [17]. According to the classification proposed by Rorke in 1983, intracranial PNET was thought to be a spectrum of disease arising from different parts of the brain [37]. Medulloblastoma, the most common intracranial PNET, is located in the posterior fossa. The diagnostic workup and treatment policy for infra- and supratentorial PNET are not clearly distinguishable. However, the prognosis in patients with SPNET is worse in comparison to those with posterior fossa PNET [4, 30]. The current study was conducted at our institute to evaluate the clinical features, treatment protocol, patterns of failure and survival outcome in 15 consecutive patients with nonpineal SPNET.
Methods
Patient selection
We performed a comprehensive analysis of patients with nonpineal SPNET undergoing treatment at our institute from January 2006 to December 2012 by retrospective chart review. Demographic features, clinical characteristics including radiological findings, surgical details, histopathological features, adjuvant treatment policy and clinical outcome were recorded in a pre-designed proforma.
Diagnostic workup
Preoperative hemogram, liver and kidney function tests, chest X-ray and contrast-enhanced magnetic resonance imaging (MRI) of the brain were done for all patients. After obtaining the final histopathology report, the patients were evaluated by the neuro-oncologist for adjuvant therapy. Postoperative contrast-enhanced MRI of the entire neuraxis and cerebrospinal fluid (CSF) cytology were done in all patients for risk categorization prior to the start of adjuvant treatment.
Surgical policy
Maximal safe resection was attempted in all patients at the Department of Neurosurgery at our institute. The extent of resection was ascertained from the surgeon’s intraoperative notes and postoperative imaging (MRI of the brain). Owing to the aggressive nature and predilection for CSF dissemination in these tumors, adjuvant therapy was administered in all patients expeditiously.
Radiotherapy policy
Adjuvant radiation was started within 4 to 6 weeks after surgery. The clinical target volume (CTV) consisted of the entire brain and spinal axis extending at least 1 cm below the termination of the thecal sac. An isotropic margin of 5 mm was given around the CTV to generate the planning target volume (PTV). Radiation was delivered by a combination of two cranial fields and 1–2 spinal fields with 6 MV X-rays. Radiation planning was done using the Eclipse treatment planning system, version 6.5 (Varian Medical Systems, Palo Alto, CA, USA). Fit patients more than 3 years old were offered craniospinal irradiation: 36 Gy/20 fractions/4 weeks to the entire neuraxis followed by a local boost of 20 Gy/10 fractions/2 weeks usually by three-dimensional conformal radiotherapy on a CL 2300 CD linear accelerator (Varian Medical System, Palo Alto, CA, USA). A boost dose of 5.4–9 Gy at 1.8 Gy per fraction was considered for isolated spinal drop metastasis. In children below 5 years of age, immobilization was achieved with the aid of general anesthesia if required. Blood counts were repeated twice every week, and toxicity charting was done weekly using the RTOG acute radiation morbidity scoring criterion. Radiotherapy was withheld, and appropriate supportive care was given for emergence of grade 3/4 toxicity.
Chemotherapy policy
Adjuvant chemotherapy was administered in all fit patients because of the high propensity of leptomeningeal dissemination and systemic recurrence. The chemotherapy regimen consisted of 12 alternating cycles of VAC (vincristine, 1.5 mg/m2 IV D1, D8 and D15 with a top dose of 2 mg; adriamycin,75 mg/m2 IV D1; cyclophosphamide, 1.2 gm/m2 IV D1 with Mesna uro-protection) and IE (ifosfamide, 1.8 gm/m2 IV D1–D5 with Mesna uro-protection; etoposide 100 mg/m2 IV D1–D5) repeated every 3 weeks. Adriamycin was replaced with actinomycin D during the sixth cycle of VAC. Chemotherapy interruption or dose reduction was done in case of emergence of grade 3/4 hematological or non-hematological toxicities.
Follow-up policy
After completion of treatment, all patients were clinically reviewed every 3 months for the first 2 years, then every 6 months from the 3rd to 5th year and annually once thereafter. Contrast-enhanced MRI of the entire neuraxis was done at the time of first follow-up, after 1 year and then only on suspicion of disease recurrence. Response assessment was done as per the modified McDonald criterion [24].
Statistical analysis
An event was defined as death (due to disease progression or treatment-related toxicity), disease progression or recurrence. EFS, RFS and OS were defined as the interval of time from the date of diagnosis to the date of any event, documented disease recurrence or death, respectively. Survival analysis was done by Kaplan-Meier product limit method. Statistical analysis was done using MedCalc software (version 11.3.0). Patients alive at last follow-up were censored.
Results (Table 1)
Patient characteristics
Fifteen patients met the study criterion. A male sex predilection was noted (male:female = 2:1). Median age at presentation was 11 years (range 3–49 years). Presenting features included vomiting in six (40 %), headache in five (33.33 %), visual deterioration in five (33.33 %), motor impairment in four (26.67 %), seizures in four (26.67 %) and scalp swelling in three (20 %) patients. Hearing loss and ataxia were noted in one patient each. Median symptom duration was 3 months. Median Karnofsky performance scale (KPS) was 80 (range: 50–90).
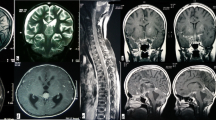
Radiology (Fig. 1)

a T2-weighted axial MRI image showing a large, lobulated, solid cystic mass involving the left parieto-occipital lobe, b T1-weighted post-gadolinium sagittal image and c coronal image show the same mass with a large cystic and solid component with heterogeneous contrast enhancement, mass effect and midline shift
Contrast-enhanced MRI of the brain revealed heterogeneous contrast-enhancing solid cystic supratentorial lesions. Tumor location was frontal in three, temporal in two, occipital in one, multilobed in five, ventricular in one, thalamic in one, midbrain in one and suprasellar in one patient. At presentation, two (13.33 %) patients had leptomeningeal dissemination.
Surgery
All patients underwent maximal safe resection. Surgical resection was gross total in six (40 %) and subtotal in eight (53.33 %) patients. Surgery was not possible in one patient. Large tumor size and extension to eloquent areas often precluded a complete surgical resection. Postoperative residuum was noted in nine patients on imaging. Three patients required the placement of a medium pressure ventriculo-peritoneal (MPVP) shunt for hydrocephalus.
Pathology (Fig. 2)

a Photomicrograph showing a highly cellular tumor with tumor cells arranged in diffuse sheets with foci of necrosis (×100); b the tumor cells have a high nucleo-cytoplasmic ratio with scant cytoplasm and indistinct cell borders, vesicular nuclei with indistinct nucleoli (×200); c the tumor cells are immunopositive for synaptophysin (cytoplasmic staining) (×200); d the tumor cells are immunonegative for GFAP (×200); e immunostaining for MIC-2 is negative (×200); f MIB-1 stain showing high proliferative activity of the tumor (labeling index >90 %) (×200)
Postoperative histopathology showed highly cellular, poorly differentiated tumors comprising small round cells with high nucleo-cytoplasmic ratios. The tumor cells were arranged in sheets. The tumors had brisk mitotic activity. All cases were immunopositive for synaptophysin, class III ß-tubulin, neuron-specific enolase (NSE) and neurofilament protein, but immunonegative for glial fibrillary acidic protein (GFAP) and MIC-2. The MIB-1 labeling index was high in all cases (median, 35 %; range, 25–80 %) indicating high proliferative activity of the tumor.
Radiation therapy
Radiation therapy was delivered in 11 (73.33 %) patients: craniospinal irradiation in 8 (36 Gy/20 fractions/4 weeks to the craniospinal axis followed by a local boost of 20 Gy/10 fractions/2 weeks) and focal RT in 3 patients. One patient progressed before initiation of radiotherapy and was lost to follow-up. Radiotherapy-related toxicities included dermatitis: grade 1 in two and grade 3 in one; CNS reaction: grade 1/2 in two; pharyngitis: grade 1 in one; neutropenia: grade 2 in two and grade 3 in one patient.
Systemic chemotherapy
Systemic chemotherapy with an alternating VAC and IE regimen was given in 13 (86.67 %) patients. Though we aimed to give a total of 12 cycles of chemotherapy with an alternating VAC/IE regimen, many of our patients struggled through the course of chemotherapy because of persistent myelosuppression after craniospinal irradiation, further compounded by adjuvant cytotoxic chemotherapy. Median number of cycles administered was six (range 1–16). Chemotherapy-related grade 3/4 toxicity included neutropenia in eight (53.33 %), thrombocytopenia in four (26.67 %), anemia in three (20 %) and mucositis in two (13.33 %) patients. Three (20 %) patients had neutropenic fever, and one of them died of sepsis. The most common sequence of treatment was surgery followed by radiotherapy and chemotherapy.
Survival analysis (Fig. 3)

Kaplan-Meier survival curves depicting (a) event-free survival and (b) overall survival in this cohort of 15 patients with SPNET
After a median follow-up of 22.6 months (mean, 24.47 months), complete response and progressive disease were noted in eight (53.33 %) and seven (46.67 %) patients, respectively. Four (26.67 %) patients died: three due to disease progression and one due to treatment-related toxicity (neutropenic sepsis). Median OS was not reached (actuarial rate of OS at 1 and 2 years: 81.8 and 72.7 %, respectively), and estimated median EFS was noted to be 4.12 years (actuarial rate of EFS at 1 and 2 years, 70.9 and 55.2 %, respectively).
Patterns of failure
Recurrence was noted in six (40 %) patients: local in three patients, leptomeningeal dissemination in two and both in one. The median time to disease recurrence was 11.12 months (mean, 20.67 months). The actuarial rate of recurrence-free survival at 1 and 2 years was 77 and 57.8 %, respectively.
Salvage treatment
Salvage therapy was offered to one patient with multiagent chemotherapy with a combination of intravenous vincristine, etoposide and carboplatin and weekly intrathecal methotrexate. The patient achieved complete response after six cycles but developed diffuse leptomeningeal disease 6 months after completion of chemotherapy. Best supportive care was given to the remaining five patients with recurrent disease because of poor general health and advanced disease.
Discussion
Hart et al. described supratentorial primitive neuroectodermal tumor (SPNET) as a poorly differentiated intracranial embryonal tumor in 1973 [17]. SPNET is an extremely rare central nervous system (CNS) tumor and constitutes 2.5 % of all pediatric brain tumors [5, 11, 17, 27]. Although histologically indistinguishable from other small round cell tumors of the brain, SPNET is characterized by its distinct aggressive clinical behavior and poorer outcome [19, 33, 37]. There is mounting evidence that SPNET and medulloblastoma (MB) have different molecular alterations and different responses to treatment [4, 5, 19, 38]. Gain of chromosome 17q is more common in MB, whereas loss of chromosome 14q is more common in SPNET [38]. Recently, Picard and colleagues reported three distinct molecular subgroups of central nervous system PNET distinguished by primitive neural (group 1), oligoneural (group 2) and mesenchymal (group 3) lineage with differential expression of cell lineage markers LIN28 and OLIG2 [34]. Patients in group 1 were predominantly females with younger age at presentation and dismal prognosis, the median survival being 0.8 years compared to 1.8 and 4.3 years in group 2 and group 3, respectively.
SPNET is commonly located within the cerebral cortex and pineal region (pinealoblastoma) [9, 36]. These tumors are histologically heterogeneous with variable amounts of glial, neuronal and ependymal differentiation [7]. SPNET commonly affects children and young adults, and the median age at presentation in our study was 11 years. Patients with SPNET present with a wide range of symptoms, which is attributable to their varying locations. Headache, vomiting and visual disturbance were the common presenting features in our study cohort. SPNET shows a high propensity of CSF dissemination (14–20 %) [28, 31]. In the present series, leptomeningeal spread was observed in two patients at presentation and another three at failure.
The management of SPNET has largely evolved based on the treatment philosophy for high-risk medulloblastoma [36]. Multimodality management comprising surgery, radiation therapy and systemic chemotherapy is essential for therapeutic success in this rare tumor [28]. Surgery is the cornerstone of management and offers rapid symptom relief and long-term disease control [42]. Variable rates of gross total resection (GTR) have been reported in the literature, ranging from 20 to 53.33 % (Table 2). Young age at diagnosis, large tumor size and extension of the tumor to eloquent areas of the brain often preclude GTR. In the available literature, the prognostic significance of achieving GTR is controversial [6, 7, 21, 22, 28]. Albright et al., in a series of 27 patients with SPNET treated with the CCG-921 protocol, showed that postoperative survival at 4 years was 40 versus 13 % in patients with a postoperative residuum of less than and more than 1.5 cm2, respectively [1]. There was a trend toward better survival in children undergoing GTR (P = 0.08) in a series of 36 patients with SPNET from the Hospital for Sick Children, Toronto, reported by Dirks et al. [7]. In the present study, GTR could only be accomplished in 40 % of patients.
Keeping in mind the natural history of the tumor, craniospinal irradiation (CSI) followed by a local boost to tumor bed is considered standard [31]. The most commonly used time-dose fractionation schedule is CSI to a dose of 36 Gy (range 18–40 Gy) followed by a local boost to 54 Gy (range 45–72 Gy) in conventional fractionation (Table 2). Reduced dose CSI (23.4 Gy) or focal RT alone has been used in very young children (1.5–3 years) in a few studies to minimize the late effects of radiation (Table 2). Paulino et al., in a series of 25 patients with SPNET, reported 5- and 10-year progression-free survival (PFS) rates of 47.1 % in patients treated with CSI compared to 12.5 and 0 %, respectively, in patients undergoing whole-brain RT (WBRT) or focal RT (P = 0.02) [31]. Failure at the untreated neuraxis site was the most common cause of progression in six out of eight patients receiving WBRT or focal RT [31]. McBride et al., in a retrospective review of 15 patients with nonpineal SPNET, observed a statistically significant difference in overall survival in patients who received upfront RT versus those who did not (P = 0.048) [28]. In our series, radiation therapy was delivered in 11 (73.33 %) patients, comprehensive craniospinal irradiation in 8 and focal RT in 3 patients.
Exploiting the differential repair capacity of normal and tumor tissues, hyperfractionated radiotherapy (HFRT) is an attractive option in this rare tumor, which is believed to be more resistant to conventional radiotherapy and chemotherapy in comparison to medullobastoma [2, 12, 27]. In various studies, CSI (31.2–40 Gy) followed by a local boost (up to 59.7–72 Gy) have been delivered using 1–1.3 Gy per fraction and 2 fractions per day, 6–8 hours apart, with a view to achieving superior tumor control by dose escalation with simultaneous minimization of late morbidities [2, 12, 27] (Table 2). The clinical outcome in these studies has been quite satisfactory with overall survival rates at 3 and 5 years in excess of 60 and 50 %, respectively (Table 2). However, this approach is quite resource intensive, and in young children who require the aid of general anesthesia or conscious sedation for immobilization, delivery of two fractions per day may be quite challenging from the nutritional point of view.
Taking a cue from average-risk medulloblastoma, Chintagumpala et al. explored the efficacy of risk-adapted treatment in patients with SPNET [5]. In a study of 16 patients with SPNET (pineal, 7; nonpineal, 9), eight average-risk patients underwent CSI to a dose of 23.4 Gy, and eight high-risk patients underwent CSI to a dose of 36 Gy in M0 disease with postoperative residuum more than 1.5 cm2 or 36–39.6 Gy in M2 disease and 39.6 Gy in M3 disease. All patients received a three-dimensional conformal boost to the tumor bed to a dose of 55.8 Gy and to metastatic sites to a dose of 50.4 Gy. After a gap of 6 weeks, all patients received four cycles of non-myeloablative high-dose chemotherapy with cisplatin, vincristine and cyclophosphamide followed by autologous stem cell rescue. After a median follow-up of 5.4 years, 12 patients were alive with a 5-year EFS rate of 75 and 60 % and 5-year OS rate of 88 and 58 % in average- and high-risk patients, respectively. This small pilot study suggests that risk-adapted craniospinal irradiation is feasible in patients with SPNET, provided they receive high-dose chemotherapy in the adjuvant setting (Table 2).
Adjuvant chemotherapy using a multidrug regimen is advocated to enhance systemic control in patients with SPNET [8, 14, 43]. In the CCG 921 trial by Cohen et al., 55 patients with SPNET after surgery were randomized to receive craniospinal radiotherapy followed by eight cycles of lomustine, vincristine and prednisone (standard treatment) or two cycles of 8-in-1 chemotherapy followed by CSI and then eight additional cycles of 8-in-1 [6]. There was no significant difference in overall survival in the two arms, and there was substantial toxicity in the more intensive 8-in-1 chemotherapy arm. In a retrospective analysis of 36 patients with SPNET treated over 25 years, Dirks et al. demonstrated a trend toward better prognosis in children receiving systemic chemotherapy [7]. In the SIOP/UKCCSG PNET 3 trial by Pizer et al., 68 patients with SPNET (pineal, 23 %; nonpineal, 77 %) received postoperative radiation to a dose of 55 Gy/33 fractions at 1.67 Gy/fraction/day (CSI-35 Gy → boost-20 Gy) [35, 40]. Pre-irradiation chemotherapy (4 cycles, alternating vincristine, etoposide, carboplatin and vincristine, etoposide, cyclophosphamide) was administered in 44 patients. The overall 5-year event-free survival (EFS) and OS for the entire cohort were 51.8 and 51.5 %, respectively. The addition of pre-irradiation chemotherapy did not favorably affect survival outcome in this study. In another study of 22 patients, Reddy et al. used eight cycles of a 6-weekly regimen of cisplatin, vincristine and lomustine after maximal safe resection and CSI with satisfactory results (5-year OS, 53 %) [36]. However, in another series of 33 patients with embryonal CNS tumor from Addenbrooke’s Hospital, Cambridge, UK, use of Packer’s regimen after surgery and CSI led to dismal results in patients with nonpineal SPNET when compared with MB (5-year OS 12 % versus 79 %; P = 0.0003) [4]. The authors opined that nonpineal SPNET, especially in teenagers and young adults, is clinically distinct from MB and resistant to Packer’s regimen and suggested the use of alternative chemotherapy (ifosfamide/temozolomide based) regimens [4]. Extrapolating from the data on the use of an alternating VAC and IE regimen in peripheral PNET and Ewing's sarcoma of bone (CCG-7881 and POG-8850 protocol), we follow alternating VAC/IE-based adjuvant chemotherapy for a total of 12 cycles after the completion of radiation therapy, and all but two patients received the aforementioned regimen in the current series [15].
Due to concern about long-term neurocognitive and neuroendocrine sequelae associated with CSI, attempts have been made to eliminate, delay or limit the use of RT (CSI) with the use of prolonged [8, 13, 25] or intensive [41] or high-dose [9, 10, 26] chemotherapy with autologous bone marrow/stem cell rescue (Table 2). Though CSI was avoided or deferred in a subset or majority of patients undergoing prolonged conventional chemotherapy, the clinical outcome was unsatisfactory with 2-year OS rates ranging from 20 to 30 % [8, 13, 25] (Table 2). In the German HIT-SKK87 and HIT-SKK92 trial involving 29 children with SPNET (pineal, 2; nonpineal, 27) <3 years old, use of methotrexate-based intensive chemotherapy led to dismal 3-year rates of overall and progression-free survival (PFS) of 17.2 and 14.9 %, respectively [41]. In this trial overall 14 patients received RT while 15 did not. This perhaps reflects the fact that even if intensive chemotherapy is given, omission of RT jeopardizes survival, and delay of RT should be limited to a maximum of 6 months in SPNET patients. On the contrary, use of postoperative high-dose chemotherapy followed by autologous bone marrow/stem cell rescue in patients with SPNET in the Head Start I and II and HIT 2000 trials led to fairly impressive results with 5-year OS ranging from 40 to 50 % [9, 10]. The majority of the long-term survivors did not receive RT as part of primary treatment (Table 2). However, needless to say that high-dose chemotherapy is resource intensive, has a formidable toxicity profile (myelosuppression, infection, bleeding, mucositis, etc.) and can lead to treatment-related mortality in 5–10 % of patients [9, 26]. In a pertinent study of late effects in 21 survivors of childhood CNS tumor (MB, 13; SPNET, 4; ependymoma, 3; atypical teratoid rhabdoid tumor, 1) treated according to the Head Start I and II protocols, after a median follow-up of 12.6 years, toxicities involving CNS, vision, hearing (grade III/IV) and dentition were noted in 67 %, 67 %, 67 % and 52 % patients, respectively [39]. Hypothyroidism and growth hormone (GH) deficiency were reported in 33 and 48 % of patients, respectively. Irradiation-free survivors (N = 10; 48 %), compared to patients who had received RT, had lower rates of hypothyroidism (0/10 versus 7/11; P = 0.004) and GH deficiency (2/10 versus 8/11; P = 0.03). No case of secondary leukemia or CNS tumor was reported in the survivors.
In summary, in spite of improvement in the treatment modalities of SPNET, the reported clinical outcome is inferior to that of medulloblastoma. The OS rates at 3 and 5 years range from 17.2 to 75 % and 14 to 88 %, respectively, in different studies (Table 2). The PFS/EFS rates at 3 and 5 years range from 0 to 63 % and 17 to 75 %, respectively, in the available literature (Table 2). Tumor recurrence may be local (6.25–60 %), leptomeningeal (0–28 %) or a combination of both (6.25–28 %) (Table 2). Occasionally there may also be distant failure. Local recurrence is the dominant pattern of failure in most studies [10, 21–23, 30, 34, 35]. Gerber et al., in their study of 26 patients, demonstrated that distant failure was more common in patients with pinealoblastoma, whereas local failure was more common in patients with nonpineal SPNET [12]. In an elegant study of 133 patients with pediatric embryonal CNS tumor from North America by Perreault et al., overall 49 (37 %) patients relapsed [32]. The majority of failures were local (79 %) in nonpineal SPNET, diffuse leptomeningeal (100 %) in pinealoblastoma and diverse in medulloblastoma (local, 27 %; distant, 35 %; diffuse leptomeningeal, 38 %). Spinal relapse was noted in 100, 51, 43 and 9 % of patients with pinealoblastoma, medulloblastoma, atypical teratoid rhabdoid tumor and nonpineal SPNET, respectively. In the present study, six (40 %) patients had recurrence: local in three, leptomeningeal in two and both in one. Median time to recurrence in our study was noted to be 11.12 months. Salvage treatment options for recurrent SPNET are limited. Re-excision and second-line systemic chemotherapy may be considered for local and distant failure, respectively.
The various prognostic factors determining improved clinical outcome in patients with SPNET include age (more than 3 years versus less than 3 years) [1, 7], M stage (M0 versus M+ disease) [1, 6, 13, 31, 36], extent of resection (complete versus incomplete resection) [1, 7, 8, 21, 25, 28, 36] and use of radiation therapy (CSI) [28, 31, 41] (Table 2). SPNETs arising in adults and children differ at the molecular level with IDH1 mutation being frequent in adults (42 %) but not in children [3, 18]. In a series of 12 adult patients with nonpineal SPNET reported by Kim et al., clinical outcome was impressive with OS and PFS at 3 years being 75 and 63 %, respectively [23]. However, the authors found no difference in clinical manifestations, radiological findings and prognosis between adults and children with this tumor [23, 44]. In another comprehensive review of 57 adult patients with SPNET, Ohba et al. reported OS rates of 38.2 and 26 % at 3 and 5 years, respectively, and recommended multimodality management consisting of gross total resection (if possible), craniospinal irradiation and systemic chemotherapy for these patients [29]. It is notable that five patients (33.33 %) in our series were adults, with three having complete response and two progressive disease at last follow-up. There was no significant difference between pediatric and adult patients on univariate analysis of EFS in our series. The impact of tumor location (pineal versus nonpineal) on clinical outcome is unclear with some studies reporting no significant difference [2, 10, 36], while some show superior outcome in patients with pinealoblastoma [6, 12, 20, 21, 35] but others the contrary [5, 9, 13, 25]. Though most single-institute or cooperative trials have grouped patients with both pineal and nonpineal SPNET, we included only patients with nonpineal SPNET in our study. At the present juncture, aggressive multimodality treatment consisting of maximal safe surgery, craniospinal irradiation and adjuvant systemic chemotherapy should be strongly considered in suitable SPNET patients with an aim to achieve an OS rate of 60–70 % at 3 years and 50–60 % at 5 years. With the use of this multimodality approach, the actuarial rates of OS and EFS at 2 years in our series are 72.7 and 55.2 %, respectively. In spite of a few limitations of our study owing to its retrospective nature, relatively short follow-up and treatment heterogeneity, overall compliance with the multimodality treatment approach was noted to be good, and clinical outcome and patterns of failure in patients in this study are in concordance with published results in the medical literature. Moreover, the effectiveness of this combined modality approach in an unselected patient population in the real-world scenario can be considered a strength of our study. Longer follow-up will no doubt lead to unfolding of late recurrence and long-term effects of treatment. More attention needs to be focused on improving treatment effectiveness by innovative study designs such as altered fractionation, radiotherapy dose escalation, use of concurrent chemotherapy along with radiotherapy, intensification of adjuvant chemotherapy, use of high-dose chemotherapy with autologous stem cell rescue and simultaneously minimizing treatment morbidity by use of novel techniques such as intensity-modulated radiation with helical tomotherapy and proton beam therapy. Keeping in mind the scarcity of health resources in developing nations, the cost-benefit ratio of any such future approach needs careful consideration.
Conclusion
Compared to medulloblastoma, the prognosis of SPNET has been historically poor with little improvement over time. Aggressive multimodality management consisting of maximal safe surgery, preferably gross total excision of tumor, followed by craniospinal irradiation and systemic chemotherapy with 6–12 cycles of an alternating regimen of VAC and IE is a reasonable treatment strategy in patients with nonpineal SPNET in a developing nation. However, in young children less than 3 years, an attempt should be made to defer radiation therapy by using prolonged or intensive chemotherapy. In the future, more attention needs to be focused on innovative study designs, quality of life issues and late effects of treatment. A better understanding of the molecular biology of this enigmatic tumor may lead to refinement of the treatment strategy and improvement of clinical outcome.
References
Albright AL, Wisoff JH, Zeltzer P, Boyett J, Rorke LB, Stanley P, Geyer JR, Milstein JM (1995) Prognostic factors in children with supratentorial (nonpineal) primitive neuroectodermal tumors. A neurosurgical perspective from the children’s cancer group. Pediatr Neurosurg 22:1–7
Allen J, Donahue B, Mehta M, Miller DC, Rorke LB, Jakacki R, Robertson P, Sposto R, Holmes E, Vezina G, Muraszko K, Puccetti D, Prados M, Chan KW (2009) A phase II study of preradiotherapy chemotherapy followed by hyperfractionated radiotherapy for newly diagnosed high-risk medulloblastoma/primitive neuroectodermal tumor: a report from the Children’s Oncology Group (CCG 9931). Int J Radiat Oncol Biol Phys 74:1006–1011
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Biswas S, Burke A, Cherian S, Williams D, Nicholson J, Horan G, Jefferies S, Williams M, Earl HM, Burnet NG, Hatcher H (2009) Non-pineal supratentorial primitive neuro-ectodermal tumors (sPNET) in teenagers and young adults: time to reconsider cisplatin based chemotherapy after cranio-spinal irradiation? Pediatr Blood Cancer 52:796–803
Chintagumpala M, Hassall T, Palmer S, Ashley D, Wallace D, Kasow K, Merchant TE, Krasin MJ, Dauser R, Boop F, Krance R, Woo S, Cheuk R, Lau C, Gilbertson R, Gajjar A (2009) A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro-Oncology 11:33–40
Cohen BH, Zeltzer PM, Boyett JM, Geyer JR, Allen JC, Finlay JL, McGuire-Cullen P, Milstein JM, Rorke LB, Stanley P, Stehbens JA, Shurin SB, Wisoff J, Stevens KR, Al A (1995) Prognostic factors and treatment results for supratentorial primitive neuroectodermaltumors in children using radiation and chemotherapy: a childrens cancer group randomized trial. J Clin Oncol 13:1687–1696
Dirks PB, Harris L, Hoffman HJ, Humphreys RP, Drake JM, Rutka JT (1996) Supratentorial primitive neuroectodermal tumors in children. J Neurooncol 29:75–84
Duffner PK, Horowitz ME, Krischer JP, Friedman HS, Burger PC, Cohen ME, Sanford RA, Mulhern RK, James HE, Freeman CR, Seidel FG, Kun LE (1993) Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328:1725–1731
Fangusaro J, Finlay J, Sposto R, Ji L, Saly M, Zacharoulis S, Asgharzadeh S, Abromowitch M, Olshefski R, Halpern S, Dubowy R, Comito M, Diez B, Kellie S, Hukin J, Rosenblum M, Dunkel I, Miller DC, Allen J, Gardner S (2008) Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the head start I and II experience. Pediatr Blood Cancer 50:312–318
Friedrich C, von Bueren AO, von Hoff K, Gerber NU, Ottensmeier H, Deinlein F, Benesch M, Kwiecien R, Pietsch T, Warmuth-Metz M, Faldum A, Kuehl J, Kortmann RD, Rutkowski S (2013) Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro-Oncology 15:224–234
Gaffney CC, Sloane JP, Bradley NJ, Bloom HJ (1985) Primitive neuroectodermal tumours of the cerebrum. Pathology and treatment. J Neurooncol 3:23–33
Gerber NU, von Hoff K, Resch A, Ottensmeier H, Kwiecien R, Faldum A, Matuschek C, Hornung D, Bremer M, Benesch M, Pietsch T, Warmuth-Metz M, Kuehl J, Rutkowski S, Kortmann RD (2014) Treatment of children with central nervous system primitive neuroectodermal tumors/pinealoblastomas in the prospective multicentric trial HIT 2000 using hyperfractionated radiation therapy followed by maintenance chemotherapy. Int J Radiat Oncol Biol Phys 89:863–871
Geyer JR, Zeltzer PM, Boyett JM, Rorke LB, Stanley P, Albright AL, Wisoff JH, Milstein JM, Allen JC, Finlay JL, Ayers GD, Shurin SB, Stevens KR, Bleyer WA (1994) Survival of infants with primitive neuroectodermal tumors or malignant ependymoma of the CNS treated with eight drugs in 1 day: a report from the childrens cancer group. J Clin Oncol 12:1607–1615
Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, Breiger D, Broxson E, Donahue B, Finlay JL, Goldwein JW, Heier LA, Johnson D, Mazewski C, Miller DC, Packer R, Puccetti D, Radcliffe J, Tao ML, Shiminski-Maher T, Children’s Cancer Group (2005) Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s cancer group. J Clin Oncol 23:7621–7631
Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers PA, Donaldson SS, Moore S, Rausen AR, Vietti TJ, Miser JS (2003) Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348:694–701
Gururangan S, McLaughlin C, Quinn J, Rich J, Reardon D, Halperin EC, Herndon J 2nd, Fuchs H, George T, Provenzale J, Watral M, McLendon RE, Friedman A, Friedman HS, Kurtzberg J, Vredenbergh J, Martin PL (2003) High-dose chemotherapy with autologous stem-cell rescue in children and adults with newly diagnosed pineoblastomas. J Clin Oncol 21:2187–2191
Hart MN, Earle KM (1973) Primitive neuroectodermal tumors of the brain in children. Cancer 32:890–897
Hayden JT, Frühwald MC, Hasselblatt M, Ellison DW, Bailey S, Clifford SC (2009) Frequent IDH1 mutations in supratentorial primitive neuroectodermal tumors (sPNET) of adults but not children. Cell Cycle 8:1806–1807
Inda MM, Perot C, Guillaud-Bataille M, Danglot G, Rey JA, Bello MJ, Fan X, Eberhart C, Zazpe I, Portillo E, Tuñón T, Martínez-Peñuela JM, Bernheim A, Castresana JS (2005) Genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermaltumours of the central nervous system. Histopathology 47:631–637
Jakacki R, Zeltzer PM, Boyett JM, Albright AL, Allen JC, Geyer JR, Rorke LB, Stanley P, Stevens KR, Wisoff J et al (1995) Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the children’s cancer group. J Clin Oncol 13:1377–1383
Jakacki RI, Burger PC, Kocak M, Boyett JM, Goldwein J, Mehta M, Packer RJ, Tarbell NJ, Pollack IF (2015) Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children’s Oncology Group. Pediatr Blood Cancer 62:776–783
Johnston DL, Keene DL, Lafay-Cousin L, Steinbok P, Sung L, Carret AS, Crooks B, Strother D, Wilson B, Odame I, Eisenstat DD, Mpofu C, Zelcer S, Huang A, Bouffet E (2007) Supratentorial primitive neuroectodermaltumors: a Canadian pediatric brain tumor consortium report. J Neurooncol 86:101–108
Kim DG, Lee DY, Paek SH, Chi JG, Choe G, Jung HW (2002) Supratentorial primitive neuroectodermal tumors in adults. J Neurooncol 60:43–52
Macdonald D, Cascino T, Schold SJ, Cairncross J (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8:1277–1280
Marec-Berard P, Jouvet A, Thiesse P, Kalifa C, Doz F, Frappaz D (2002) Supratentorial embryonal tumors in children under 5 years of age: an SFOP study of treatment with postoperative chemotherapy alone. Med Pediatr Oncol 38:83–90
Mason WP, Grovas A, Halpern S, Dunkel IJ, Garvin J, Heller G, Rosenblum M, Gardner S, Lyden D, Sands S, Puccetti D, Lindsley K, Merchant TE, O’Malley B, Bayer L, Petriccione MM, Allen J, Finlay JL (1998) Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol 16:210–221
Massimino M, Gandola L, Spreafico F, Luksch R, Collini P, Giangaspero F, Simonetti F, Casanova M, Cefalo G, Pignoli E, Ferrari A, Terenziani M, Podda M, Meazza C, Polastri D, Poggi G, Ravagnani F, Fossati-Bellani F (2006) Supratentorial primitive neuroectodermal tumors (SPNET) in children: a prospective experience with adjuvant intensive chemotherapy and hyperfractionated accelerated radiotherapy. Int J Radiat Oncol Biol Phys 64:1031–1037
McBride SM, Daganzo SM, Banerjee A, Gupta N, Lamborn KR, Prados MD, Berger MS, Wara WM, Haas-Kogan DA (2008) Radiation is an important component of multimodality therapy for pediatric non-pineal supratentorial primitive neuroectodermal tumors. Int J Radiat Oncol Biol Phys 72:1319–1323
Ohba S, Yoshida K, Hirose Y, Ikeda E, Kawase T (2008) A supratentorial primitive neuroectodermal tumor in an adult: a case report and review of the literature. J Neurooncol 86:217–224
Paulino AC, Melian E (1999) Medulloblastoma and supratentorial primitive neuroectodermal tumors: an institutional experience. Cancer 86:142–148
Paulino AC, Cha DT, Barker JL Jr, Lo S, Manera RB (2004) Patterns of failure in relation to radiotherapy fields in supratentorial primitive neuroectodermal tumor. Int J Radiat Oncol Biol Phys 58:1171–1176
Perreault LRM, Carret AS, Zhang G, Hershon L, Décarie JC, Yeom K, Vogel H, Fisher PG, Partap S (2013) Relapse pattern in pediatric embryonal central nervous system tumors. J Neurooncol 115:209–215
Pfister S, Remke M, Toedt G, Werft W, Benner A, Mendrzyk F, Wittmann A, Devens F, von Hoff K, Rutkowski S, Kulozik A, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2007) Supratentorial primitive neuroectodermal tumors of the central nervous system frequently harbor deletions of the CDKN2A locus and other genomic aberrations distinct from medulloblastomas. Genes Chromosom Cancer 46:839–851
Picard D, Miller S, Hawkins CE, Bouffet E, Rogers HA, Chan TS, Kim SK, Ra YS, Fangusaro J, Korshunov A, Toledano H, Nakamura H, Hayden JT, Chan J, Lafay-Cousin L, Hu P, Fan X, Muraszko KM, Pomeroy SL, Lau CC, Ng HK, Jones C, Van Meter T, Clifford SC, Eberhart C, Gajjar A, Pfister SM, Grundy RG, Huang A (2012) Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13:838–848
Pizer BL, Weston CL, Robinson KJ, Ellison DW, Ironside J, Saran F, Lashford LS, Tait D, Lucraft H, Walker DA, Bailey CC, Taylor RE (2006) Analysis of patients with supratentorial primitive neuro-ectodermal tumors entered into the SIOP/UKCCSG PNET 3 study. Eur J Cancer 42:1120–1128
Reddy AT, Janss AJ, Phillips PC, Weiss HL, Packer RJ (2000) Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation and chemotherapy. Cancer 88:2189–2193
Rorke LB (1983) The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol 42:1–15
Russo C, Pellarin M, Tingby O, Bollen AW, Lamborn KR, Mohapatra G, Collins VP, Feuerstein BG (1999) Comparative genomic hybridization in patients with supratentorial and infratentorial primitive neuroectodermaltumors. Cancer 86:331–339
Saha A, Salley CG, Saigal P, Rolnitzky L, Goldberg J, Scott S, Olshefski R, Hukin J, Sands SA, Finlay J, Gardner SL (2014) Late effects in survivors of childhood CNS tumors treated on Head Start I and II protocols. Pediatr Blood Cancer 61:1644–1652
Taylor RE, Donachie P, Weston C, Robinson K, Lucraft H, Saran F, Ellison DW, Ironside J, Walker DA, Pizer BL (2009) Impact of radiotherapy parameters on outcome for patients with supratentorial primitive neuro-ectodermal tumours entered into the SIOP/UKCCSG PNET 3 study. Radiother Oncol 92:83–88
Timmermann B, Kortmann RD, Ku¨hl J, Rutkowski S, Meisner C, Pietsch T, Deinlein F, Urban C, Warmuth-Metz M, Bamberg M (2006) Role of radiotherapy in supratentorial primitive neuroectodermaltumor in young children: results of the German HIT-SKK87 and HIT-SKK92 trials. J Clin Oncol 24:1554–1560
Tomita T (1998) Neurosurgical perspectives in pediatric neurooncology. Childs Nerv Syst 14:94–96
White L, Johnston H, Jones R, Mameghan H, Nayanar V, McWhirter W, Kellie S, Waters K, Toogood I (1993) Postoperative chemotherapy without radiation in young children with malignant non-astrocytic brain tumours: a report from the Australia and New Zealand childhood cancer study group (ANZCCSG). Cancer Chemother Pharmacol 32:403–406
Yang HJ, Nam DH, Wang KC, Kim YM, Chi JG, Cho BK (1999) Supratentorial primitive neuroectodermal tumor in children: clinical features, treatment outcome and prognostic factors. Childs Nerv Syst 15:377–383
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presentation
Part of the data was accepted for a poster presentation at the 4th quadrennial meeting of the World Federation of Neuro-Oncology in San Francisco, CA, USA, November 2013. Neuro Oncol 2013; 15 (suppl3): iii79.
Rights and permissions
About this article

Cite this article
Biswas, A., Mallick, S., Purkait, S. et al. Treatment outcome and patterns of failure in patients of non-pineal supratentorial primitive neuroectodermal tumor: review of literature and clinical experience form a regional cancer center in north India. Acta Neurochir 157, 1251–1266 (2015). https://doi.org/10.1007/s00701-015-2444-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-015-2444-2