
Abstract
Terpenes are a multifarious group of secondary compounds present throughout the living world that function primarily in defence, or otherwise in regulating interactions between an organism and its environment. Terpene synthases (TPS) are a mid-sized gene family whose diversity and make-up reflects a plant’s ecological requirements and unique adaptive history. Here we catalogue TPS in Melaleuca alternifolia and examine lineage-specific expansion in TPS relative to other sequenced Myrtaceae. Overall, far fewer (37) putative TPS genes were identified in M. alternifolia compared with Eucalyptus grandis (113) and E. globulus (106). The number of genes in clade TPS-b1 (12), which encode enzymes that produce cyclic monoterpenes, was proportionally larger in M. alternifolia than in any other well-characterised plant. Relative to E. grandis, the isoprene-/ocimene-producing TPS-b2 clade in M. alternifolia tended to be proportionally smaller. This suggested there may be lineage-specific subfamily change in Melaleuca relative to other sequenced Myrtaceae, perhaps as a consequence of its semi-aquatic evolutionary history.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Terpenes are volatile, often aromatic hydrocarbon-based natural compounds produced by plants, fungi, bacteria and some insects, some of which play a role in primary metabolism but many of which are secondary metabolites (Toyomasu et al. 2007; Chen et al. 2011; Yamada et al. 2015). They are found in the essential oils, resins and other tissues of plants and are believed to increase fitness in a variety of complex ways, including deterring or attracting insects and other herbivorous or pollinating organisms, resisting fungal or bacterial infection (phytoalexins) or by acting as allelochemicals (Külheim et al. 2015). Isoprenes, for example, appear to alleviate heat stress (Behnke et al. 2007), perhaps by stabilising plant membranes or acting as antioxidants (Penuelas et al. 2005); ocimenes have been implicated in defence against insect herbivory (Navia-Giné et al. 2009; Shimoda et al. 2012). The biosynthetic pathways of terpenes are well understood, and genes for terpene synthases (TPSs—enzymes that catalyse the terminal step of terpene structural modification from 5-carbon isoprene subunits) have already been well described in plants such as Arabidopsis (Herde et al. 2008) and Eucalyptus (Keszei et al. 2010a, b).
TPS in plants typically exist as a mid-sized gene family (Chen et al. 2011) but can range in number from 1 in Physcomitrella patens (a bryophyte) to 113 in Eucalyptus grandis, with larger gene families tending to found in some woody perennials because of the key role of terpenes in defence over their long lifespans (Chen et al. 2011; Kulheim et al. 2015). Studies of the genome organisation of TPS show patterns of clustering into subfamilies at locations in the genome (e.g. Tuskan et al. 2006; Kulheim et al. 2015). This mechanism of gene family evolution is consistent with rounds of gene duplication (Cannon et al. 2004), whereby sections of chromosomes are duplicated in uneven crossing over events or by the action of transposable elements. Gene duplication is an important source of genetic variation, and duplications account for a large proportion of genes in eukaryotic genomes (Pierce 2012). When a single gene is duplicated and inserted close to the original, it is termed a local or tandem duplication (TD; Cannon et al. 2004).
As with other gene families involved in adaptive responses, expansion or contraction in gene family size for TPS is thought to occur in response to the nature of the stress (i.e. biotic or abiotic) which appears to influence the magnitude of expansion (Hanada et al. 2008). Lespinet et al. (2002) report that lineage-specific expansions of gene families resulting from retained TDs are very frequently expansions of genes involved in stress response, but it is not clear which type of stress has a stronger relationship with TDs. As an expansion in one orthologous group (OG) in response to an adaptive force acting on one species is often mirrored by a contraction of the same OG in a related but geographically separate species, lineage-specific gene family size variation leaves different genomic signatures for different adaptive histories (Blanc and Wolfe 2004).
A prominent feature of TPS enzymes is that they yield multiple products, with as many as 52 different terpenes being reported from one enzyme (Steele et al. 1998). The Myrtaceae family is notable among the plant families of southern hemisphere origins for its number of essential oil-rich taxa and the abundance of TPS genes in some species (Webb et al. 2014; Külheim et al. 2015).
Several eucalypts including Eucalyptus sp. and Corymbia citriodora, as well as Melaleuca sp. are grown commercially for terpene-rich essential oil. Among the Melaleuca, Melaleuca alternifolia (Maiden and Betche) Cheel is the most important for essential oil production because of the proven antimicrobial activity of a major constituent, terpinen-4-ol (Baker 1999; Morcia et al. 2012). Because of its commercial importance, it is arguably the best studied of any Myrtaceae in terms of terpene chemistry, biochemistry and genetics. Attempts have been made to identify genes underlying biosynthesis of commercially important terpene components and assign function (Shelton et al. 2002, 2004a, b; Keszei et al. 2010b, unreviewed RIRDC report; Webb et al. 2013, 2014) and regulation of oil yield (Webb et al. 2013), but as yet only a single candidate TPS has been reported for this species (Shelton et al. 2004a; Sharkey et al. 2005).
Here we catalogue the TPS genes identified in a draft genome sequence of Melaleuca alternifolia. We conduct comparative analysis of the TPS gene family with other sequenced Myrtaceae, including the reference Myrtaceae, Eucalyptus grandis (Grattapaglia et al. 2012; Myburg et al. 2014; Kulheim et al. 2015). We find there are comparatively few TPS in M. alternifolia relative to other woody perennials, but there is a tendency towards over-representation of the TPS-b1 clade of cyclic monoterpene synthases and under-representation of the TPS-b2 clade, a subfamily of isoprene/ocimene synthase gene class, relative to other sequenced Myrtaceae.
Materials and methods
Genome sequencing
A draft genomic sequence for the reference genotype SCU01 of Melaleuca alternifolia was generated using short read Illumina sequence data (See Online Resource 1 for details of results and methodology). This individual has chemotype 4 terpene chemistry (high 1,8-cineole and intermediate terpinen-4-ol) and was clonally replicated and archived in a germplasm resource collection located at the Lismore campus of SCU (Shepherd et al. 2015).
Sequencing was performed on a Hiseq 2000 (Illumina) at the Australian Genome Research Facility. In brief, a total of 100 Gb of high-quality paired-end 100-bp-long sequence reads were generated by an Illumina Hiseq to give approximately 141 X genome coverage based on a cytological estimate of 710 Mb (see Online Resource 2). Raw sequencing reads were trimmed to remove low-quality bases and adaptor sequences. Reads in FASTQ format were first checked for quality using FASTQC (Andrews 2015), followed by removal of adapter sequences, poly-N stretches and low-quality (Phred score <20) reads using the BBDuck module of the BBMap software package (version 34_90; http://sourceforge.net/projects/bbmap). A draft assembly of M. alternifolia was constructed using the CLC de novo assembler (CLC Bio, Aarhus, Denmark). The draft genome comprised a total of 221,396 contigs with total length of 356 Mb and an N50 of 8778 bp.
Gene annotation with the Maker pipeline version v2.31.8 (Cantarel et al. 2008) produced 33,184 draft gene models with an annotation edit distance of >0.35. Analysis of single copy gene coverage using the BUSCO method (Simão et al. 2015) predicted 90% of single copy genes (80% complete, 10% fragmented) captured in this set of contigs (data not shown). To check Maker’s efficacy, tBLASTn was used against the M. alternifolia genome assembly to explore the presence of TPS genes outside of Maker gene models (amino acid queries from Kulheim et al. 2015, see Online Resource 3). Two query sequences (TPSb line 1 & TPSf line 2) returned no hits. Hits to all other queries (116 in total) were associated (overlapping or contained within) with gene models predicted by Maker (see Online Resource 4 for tabulated results). This suggests that the pipeline, which used protein sequence evidence from Eucalyptus grandis, Corymbia citriodora and Vitus sp. to draw gene model predictions, is at least as effective as a straight homology search, having search parameters relaxed enough to allow for some missing consensus sequences and using multiple lines of evidence.
Mining the genome
Methods in a study by Külheim et al. (2015) of TPS genes in E. grandis served as a template. Using known conserved protein regions of 6 TPS subfamilies as BLAST queries (CoGe BLAST (Lyons et al. 2008) and NCBI BLAST+, using default parameters), searches were performed on the Melaleuca v1 genome assembly.
To establish whether the conserved domains (CDs) used for mining the E. grandis genome were suitable for locating TPS genes and confidently assigning subfamilies in M. alternifolia, one CD from each subfamily was BLASTed to both genomes, and the highest e-values for each search recorded. E-values for both species were indeed comparable in significance (for tabulated data see Online Resource 5), indicating that queries used to mine the well-studied E. grandis reference Myrtaceae genome are applicable to M. alternifolia.
To gather a broad pool of candidates, a cut-off e-value of 1e-08 was used to select the highest hits for each subfamily query (TPS-a, -b, -c, -e, -f and -g) to the M. alternifolia assembly. This cut-off was established when it became apparent that any hits with e-values less significant than 1e-10 invariably appeared in multiple search results, indicating that the subfamily-specific sensitivity of searches tapered off below that point. 1e-08 was chosen as a conservative value in the event that some e-values of relevant gene models happened to fall below 1e-10.
The pool of candidate gene models returned by these searches was sorted by subfamily and then structurally analysed using Gevo (https://genomevolution.org/coge/Gevo.pl; Lyons and Freeling 2008) to ascertain exon number (which varies depending upon subfamily; Külheim et al. 2015), and FeatView (https://genomevolution.org/coge/FeatView.pl) to ascertain number and placement of stop codons. Models were given a ranking according to a modified version of Külheim’s system, which is as follows: 1 = full length, no premature stop codons; 2 = full length, up to 2 stop codons; 3 = full length, no stop codon; 4 = pseudogenes, more than 2 stop codons; 5 = partial gene. (Ultimately, all classes of gene were included in the phylogeny, as incomplete genes could have been truncated simply by being part of a very short contig.)
Using ChloroP 1.1 (http://www.cbs.dtu.dk/services/ChloroP/) and PCLR release 0.9 (http://www.andrewschein.com/cgi-bin/pclr/pclr.cgi) (Schein et al. 2001), models were analysed to detect the presence of chloroplast transit peptide sequences (cTPs) (Emanuelsson et al. 1999). As all but the sesquiterpenes (C15) are produced in the chloroplast (Külheim et al. 2015), most TPS genes should contain a cTP.
Phylogeny
In order to replicate as closely as possible Külheim’s phylogeny methods, a test run was performed using only the 113 E. grandis TPS amino acid sequences published with the 2015 paper. Using PhyML 3.0 (http://phylogeny.lirmm.fr; Dereeper et al. 2008), a ClustalW alignment was constructed from the 113 sequences. Gblocks curation was skipped, as the analysis returned by a curated pipeline did not satisfactorily resolve some subfamilies (for example, TPS-e appeared as a clade flanked either side by TPS-c genes).
As per Külheim et al. (2015), the Jones–Taylor–Thornton amino acid substitution model was used to create a maximum-likelihood phylogenetic tree file (.tree) with 100 bootstrapped replicates, and the resulting file was imported to FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/; Rambaut 2014) for visualisation and editing. The tree (Online Resource 6) was manually rooted from the node at which types I and III (i.e. subfamilies c, e, f and a, b, g) diverge.
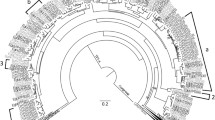
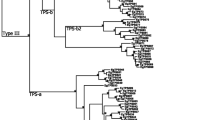
As the phylogenetic tree that resulted from using the above settings showed very high structural similarity to that of Külheim et al. (2015), the same settings were applied using the set of 113 E. grandis TPS genes plus the 37 M. alternifolia candidate gene models identified using BLAST, as well as the coding sequence for the putative monoterpene synthase transcript obtained by Shelton et al. (2004a, b; GenBank accession AY279379.1). The alignment for this phylogeny can be found in Online Resource 7. A tree was constructed as outlined above; Fig. 1 is one maximum-likelihood tree, which shows average numbers of amino acid substitutions per branch as branch length relative to the scale.

Phylogeny of 37 Melaleuca alternifolia putative TPS genes with 113 Eucalyptus grandis TPS genes from Külheim et al. (2015) and 1 M. alternifolia putative monoterpene synthase from Shelton et al. (2004a, b). Melaleuca alternifolia genes are indicated by a black dot. Scale = average number of amino acid substitutions per branch (JPEG produced using Figtree v1.4.2. And GIMP)
Results
Putative TPS genes and subfamily proportions
Thirty-seven candidate TPS genes with high similarity to conserved TPS regions were identified in the Melaleuca alternifolia genome (Table 1; Fig. 2; all gene models are listed in Online Resource 8; .fasta files of 37 amino acid sequences are attached as Online Resource 9).

Proportion of TPS gene subfamilies found in 12 plant species as listed in Table 1. Melaleuca alternifolia contains the highest proportion of TPS-b1 genes. Gene proportions in M. alternifolia do not differ significantly from those in Eucalyptus grandis (χ 2 = 1.74; χcrit = 12.59; p = 0.05). (JPEG produced using LibreOffice Calc and GIMP)
Fourteen genes clustered with subfamily TPS-a, which produce sesquiterpenes (C15); twelve with TPS-b1, which produce cyclic monoterpenes (C10, e.g. sabinene hydrate and 1,8-cineole); two with TPS-b2, which produce isoprenes and ocimenes (C5, C10); one with TPS-c, which produce diterpenes (C20); one with TPS-e, which produce mono-, sesqui- and diterpenes; three with TPS-f, which also produce mono-, sesqui- and diterpenes; and four with TPS-g, which predominantly produce acyclic mono-, sesqui- and diterpenes.
Of all well-studied plants represented in Table 1 and Fig. 2, M. alternifolia has the highest number of TPS-b1 genes as a proportion of the total number of TPS genes: 32.4%, compared with Populus trichocarpa, the next highest at 31.2%. TPS gene subfamily proportions do not differ significantly between M. alternifolia and E. grandis (χ 2 = 1.74; χcrit = 12.59; p = 0.05), although tea tree has a proportionally larger set of TPS-b1 (cyclic monoterpene) genes and a smaller set of TPS-b2 (isoprene/ocimene) genes. However, differences in subfamily proportions between M. alternifolia and both P. trichocarpa (a well-characterised woody dicot) and A. thaliana (a well-characterised herbaceous annual) were significant: χ2 = 26.85 and 36.08, respectively.
Transit peptides
Only five M. alternifolia genes from TPS subfamilies, -a (1 gene), -b1 (2), -b2 (1) and -g (1), were predicted by ChloroP 1.1 to contain cTPs. For context, the 113 E. grandis genes from Külheim et al. (2015) were run through ChloroP 1.1, which found six genes from subfamilies -a (4), -b (1) and -e (1) with cTPs. (cTP-containing genes from both species are listed in Online Resource 10.) TPS-a genes with a predicted transit peptide were compared between M. alternifolia and E. grandis. (The sole predicted cTP-containing TPS-b gene from E. grandis, Eucgr.K00875.1.v2.0, was found to be a very small, incomplete gene model, leaving only TPS-a in common between the two species.) Sequence identity between these TPS-a genes was between 70.1% (MelG016248 to Eucgr.H04978) and 82.3% (MelG016248 to Eucgr.F03396).
Interestingly, results from analysis of the same 37 gene models using PCLR r0.9 returned the same five models as predicted by ChloroP (see Online Resource 11), with no others predicted to contain chloroplast transit peptides.
Sequence identity between predicted cTP-containing TPS-a genes from both species did not greatly exceed that between TPS-a genes not predicted to contain a cTP (70.1–82.3% for genes with predicted cTPs, compared to 65.6–79.1% for those without, calculated by comparing 6 randomly selected non-cTP E. grandis TPS-a genes with MelG016248, the only M. alternifolia TPS-a gene predicted to contain a cTP). A BLAST search of the Eucalyptus grandis BRASUZ1 (Phytozome unmasked v2) genome assembly using the amino acid sequence of MelG016248 did return hits to 4 of 6 E. grandis predicted cTP-containing TPS-a genes (Eucgr.D01103, Eucgr.E00419, Eucgr.F03396, and Eucgr.H04978). However, these hits ranged from HSP #88 to #23, with many other genes returning higher scores, making it unlikely that these cTP-predicted genes from both species are orthologues.
Phylogeny
A foundation for comparative analysis was established by replicating the Külheim et al. (2015) phylogenetic tree for E. grandis. Our tree had a high degree of resemblance with that of Külheim et al. (2015) (see Online Resource 12 for tree format file), with all subfamilies resolving into clades of identical size and structure.
Inclusion of the 37 M. alternifolia candidates, however, induced some repositioning of clades (Fig. 1; see Online Resource 13 for .tree file). For example, resolution was lost in the splitting within type I subfamilies, with TPS-f appearing as a sister group to both -c and -e (in the E. grandis phylogeny, -c split off first, followed by -e and then -f). However, in the tree containing only E. grandis genes, TPS-g was a sister to the greater -b group (bootstrap at g/b node = 0.53), and the phylogeny that includes both species showed -g as a sister to -a. The inclusion of a set of genes from a different (albeit closely related) species therefore reduced certainty in the branching order of TPS subfamily clades.
The TPS-b1 gene MelG017535 showed very high divergence (as represented by branch length in Fig. 1) from the other genes in its clade. When an alignment and phylogeny were produced using only the 37 M. alternifolia genes (tree not included in this report), MelG017535 showed a similarly high divergence from other TPS-b1 genes. The gene has 6 exons—1 fewer than the usual 7 observed by Külheim et al.
Finally, the M. alternifolia mRNA sequenced and classified as a putative monoterpene synthase persistently clustered not with the TPS-b1 cyclic monoterpene subfamily, as originally proposed by Shelton et al. (2004a, b), but with the TPS-b2 isoprene/ocimene subfamily (ISPS). In addition, this mRNA sequence had 100% sequence identity to one gene model in the M. alternifolia assembly, MelG010433.
Discussion
Putative TPS genes and subfamily proportions
Given the BUSCO gene coverage estimate of 90%, it is probable that there are slightly more (41) than 37 TPS genes in the Melaleuca alternifolia genome than inferred. Refinements to the genome assembly using data derived from further sequencing may bear this out. However, in sequencing a genome as highly heterozygous as M. alternifolia, there is a chance that both alleles from one locus may be incorrectly assigned to different loci, which would appear to increase the number of paralogues on the assembly.
From the much lower number of putative TPS genes found in M. alternifolia compared to Eucalyptus grandis (37 versus 113), results imply that evolutionary forces have acted differentially upon the two lineages since they diverged. Although there are far fewer TPS genes in M. alternifolia overall, all subfamilies were nonetheless represented. TPS-c is conserved in land plants and is thought to represent the base of the TPS tree, originating as a diterpene synthase-producing gibberellin (regulatory plant hormone) precursors (Yamaguchi 2008). TPS-e and -f—conserved in vascular plants—are also linked to hormone production, sharing a common progenitor gene coding for an ent-kaurene synthase, also a gibberellin precursor (Chen et al. 2011). In contrast, TPS-a, -b and -g are angiosperm specific, and their products (mono-, sesqui- and diterpenes) have been characterised as playing ecological rather than primary metabolic or regulatory roles (Chen et al. 2011). A salient question is whether this low number of “ecological” TPS genes in M. alternifolia compared to E. grandis represents a reduction, or the retention of an ancestral state.
Orthologous pairing has been observed in most of the TPS genes in E. grandis, with large genomic clusters consisting of both functional and pseudogenes (Külheim et al. 2015) pointing to a proliferation of gene duplication events. Thornhill et al. (2015) report an estimated divergence of the genera Melaleuca and Eucalyptus at ~68 million years ago and that the closest sister tribe to the Melaleucaceae, the monotypic Osbornieae (divergence ~56 million years ago), is the only member of Myrtaceae to occur in a mangrove growth form and habitat. This suggests the existence of a basal estuarine or riparian progenitor of these tribes between 68 and 56 million years ago.
Sharkey et al. (2013) functionally characterised an isoprene synthase gene from E. globulus (EglobTPS106; GenBank AB266390.1) that is almost identical (99.6%) to the E. grandis gene EgranTPS084 (Eucgr.K00881; GenBank XM_010037321), the single E. grandis TPS-b2 gene that fulfils the criteria for isoprene synthases outlined in the 2013 Sharkey paper. The remaining 8 TPS-b2 genes are putative ocimene synthases (or of unknown function). In M. alternifolia, 2 putative TPS-b2 genes were identified by this study, one of which, MelG010433, appears to code for the mRNA transcript described by Shelton et al. (2004a, b) and functionally characterised as an ISPS by Sharkey et al. (2005). The other M. alternifolia TPS-b2 gene, MelG013034, lacks the isoprene synthase-specific amino acids and may be considered a putative ocimene synthase until it is functionally characterised. Thus, a breakdown of TPS-b2 for E. grandis is 1 isoprene, 8 ocimene, whereas for M. alternifolia it is 1 isoprene, 1 ocimene.
Transcripts encoding ocimene synthases accumulate in leaves in response to insect herbivory (Navia-Giné et al. 2009). (E)-β-ocimene appears to play a role in attracting the insect predators of herbivorous spider mites (Shimoda et al. 2012), which occur in Australia (Wilson et al. 1996). That M. alternifolia possesses only a single putative ocimene synthase gene, compared to E. grandis’ 8, suggests either that tea tree has evolved other strategies to deter herbivores or that pressures imposed by herbivory differ in magnitude or variety from those undergone by the eucalypts.
In addition, the eucalypts appear to have a proportionally smaller TPS-b1 subfamily than M. alternifolia. TPS subfamily proportions observed in Corymbia citriodora subsp. variegata tend to mirror Eucalyptus sp. ratios: a proportionally larger TPS-b2 relative to TPS-b1 (cyclic monoterpene synthases). This suggests proportionally higher representation of the TPS-b2 may be a feature of the eucalypt group more broadly, reflecting either their higher degree of relatedness or their more similar ecological history.
Conversely, the subfamily TPS-b1 is proportionally larger in M. alternifolia than in any representative plant (dicot, monocot or moss) in Fig. 2, suggesting that duplicate retention or lineage-specific gene family expansion in this subfamily has been an important adaptation in tea tree. Cyclic monoterpenes have been shown to increase membrane permeability of fungal hyphae, effectively inhibiting growth of fungal plant pathogens (Tao et al. 2014). They have also been shown to inhibit the action of bacterial polygalacturonase enzymes, which phytopathogenic bacteria use to break down the pectin of plant cell walls (Rasoul et al. 2012). Keszei et al. (2010b, unreviewed RIRDC report) hypothesise that the ancestral form of the TPS-b1 enzyme for both Melaleuca and Eucalyptus was one responsible for cineole biosynthesis. 1,8-cineole has been shown to inhibit the growth of gram-positive and gram-negative bacteria, and yeasts (Silva et al. 2011).
Given the warm, subtropical habitat of tea tree’s evolution, it is unsurprising that an arsenal of antimicrobial secondary metabolites such as cyclic monoterpenes should have been selected for. That at least two of the TPS-b1 genes appear to be the result of tandem duplication raises the possibility that biotic stress may have stimulated the expansion of this TPS subfamily. Barlow (1988) suggested that both Melaleuca and Eucalyptus may both have had their origins at rainforest margins, from whence they differentiated—Melaleuca as a seasonally drowned habitat specialist and Eucalyptus as a coloniser of low-nutrient, seasonally drier soils.
Transit peptides
The 113 E. grandis TPS genes identified by Külheim et al. (2015) are putatively functional based on RNA expression data from seven tissue types. As listed in Table 1, E. grandis has at least 38 genes that do not encode cytosol-destined sesquiterpene synthases but do encode plastid-destined TPS enzymes of other classes (from subfamilies -b1, -b2 and -c). Thus, we should expect at least 38 E. grandis genes with predicted cTPs. That ChloroP 1.1 predicted only six of these indicates that such an analysis as applied to M. alternifolia may be erroneous. Therefore, the cTP data returned by ChloroP 1.1 analysis should be regarded with caution. However, that both ChloroP and PCLR predicted cTPs in the same 5 M. alternifolia gene models despite the programs’ differing systems of prediction (neural network versus principal component analysis, respectively) adds another line of evidence to the putatively functional status of these 5 genes.
In a review of plastid transit peptides, Bruce (2001) noted that their “extreme diversity in sequence and evolution” means that they are still poorly characterised. It remains possible that the ChloroP 1.1 and PCLR r0.9 software were unable to detect many of the cTPs of TPS genes in M. alternifolia and E. grandis.
Phylogeny
Minor differences in some bootstrap values between the model phylogeny of Külheim et al. (2015) and the one in this study may have been the result of unreleased manual adjustments to the alignment performed by the authors of the 2015 study, or simply from slight variation in the 100 bootstrapped replicas used to construct the final consensus tree. Additionally, joint confidence (i.e. overall confidence incorporating the bootstrap values of all nodes) in large trees is inescapably low (Soltis and Soltis 2003). In any case, the phylogenetic trees produced in this study possess nodes with bootstrap values of <80% in similar numbers to the trees of Külheim et al., which illustrates fundamental uncertainties in the relationships between TPS subfamilies. It is tempting to view a phylogeny with high bootstrap values as being directly reflective of the actual relationships between loci. However, as Felsenstein (1985) notes, “Bootstrapping provides us with a confidence interval within which is contained not the true phylogeny, but the phylogeny that would be estimated upon repeated sampling of many characters from the underlying pool of characters”. In other words, a bootstrap value indicates only that the analysis returned the same result many times. From this, we must be careful of confidently inferring actual evolutionary relationships.
Confidence in the finer grouping of individual loci was much higher than for the broader relationships between TPS clades, both in the phylogenetic tree produced by Külheim et al. (2015) and in the two trees produced for this study (with and without M. alternifolia genes). However, the inferred relationships between TPS subfamilies mostly mirror those found by Chen et al. (2011) in a phylogeny of putative full-length TPS genes from 7 sequenced plant genomes and representative characterised gymnosperm TPS sequences. Slight differences lie in the splitting of type I (TPS-c, -e and -f) clade and in the order of branching within type III (TPS-a, -b and -g). For the purposes of assigning TPS subfamilies to gene models, however, the phylogeny produced in this study was deemed adequate.
The 2011 study by Chen et al. characterised clades TPS-a, -b and -g as encoding enzymes involved in ecological interactions rather than primary metabolism or hormonal regulation. These three subfamilies, which show considerable divergence in sequence to the other TPS clades, contain the highest number of putative TPS genes in M. alternifolia (14, 14 and 4 genes, respectively) and together make up 32 of the 37 genes identified in this study. The remaining 5 genes from TPS-c, -e and dicot-specific TPS-f (1,1 and 3 genes) are, based on the characterisation of Chen et al., likely to encode enzymes that produce plant hormone precursors.
The long branch of TPS-b1 gene MelG017535 suggests high divergence from the other genes in that clade. However, its lack of a seventh exon compared to other TPS-b1 gene models could be due to the inclusion of an intron, or fusion with another gene. If the striking difference in sequence and single lost exon are not artefacts of sequencing or errors in gene model prediction, this gene, once verified, warrants further investigation as a potential new subtype of TPS-b1.
Gene model MelG010433, which is identical in sequence to the mRNA studied and classified as a TPS-b1 monoterpene synthase gene by Shelton et al. (2004a, b), showed a tendency to cluster with TPS-b2 rather than TPS-b1 genes. This is supported by Sharkey et al. (2005), who functionally characterised this transcript as an ISPS, and by Keszei et al. (2010a, b), who also concluded that the sequence codes for an ISPS in TPS-b2.
Conclusion
This study provides crucial baseline estimates for TPS gene numbers and subfamilies in M. alternifolia. This information will be important in further elucidation of the tea tree’s evolutionary history, the broader study of gene family evolution, and in understanding in greater detail the ecological functions of terpenes in the family Myrtaceae.
Change history
05 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00606-021-01740-9
References
Andrews S (2015) FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 14 May 2016
Baker G (1999) Tea tree breeding. In: Southwell I, Lowe R (eds) Tea tree: the genus Melaleuca. Harwood Academic Publishers, Amsterdam, pp 135–154
Barlow BA (1988) Patterns of differentiation in tropical species of Melaleuca L. (Myrtaceae), pp. 239–247 in The ecology of Australia’s wet tropics: proceedings of a symposium held at the University of Queensland. In: R. L. Kitching. Surrey Beatty & Sons for Ecol Soc Aust procite:01edfb08-aef9-4a6f-bf47-0127cb4ffcde
Behnke K, Ehlting B, Teuber M, Bauerfeind M, Louis S, Hänsch R, Schnitzler JP (2007) Transgenic, non-isoprene emitting poplars don’t like it hot. Pl J 51:485–499. doi:10.1111/j.1365-313X.2007.03157.x
Blanc G, Wolfe KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Pl Cell 16:1667–1678. doi:10.1105/tpc.021345
Bruce BD (2001) The paradox of plastid transit peptides: conservation of function despite divergence in primary structure. BBA Molec Cell Res 1541:2–21. doi:10.1016/S0167-4889(01)00149-5
Cannon SB, Mitra A, Baumgarten A, Young ND, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Pl Biol 4:1. doi:10.1186/1471-2229-4-10
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Yandell M (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18:188–196. doi:10.1101/gr.6743907
Chen F, Tholl D, Bohlmann J, Pichersky E (2011) The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Pl J 66:212–229. doi:10.1111/j.1365-313X.2011.04520.x
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucl Acids Res 36(Web Server issue):W465–W469. doi:10.1093/nar/gkn180
Emanuelsson O, Nielsen H, Von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8:978–984. doi:10.1110/ps.8.5.978
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi:10.2307/2408678
Grattapaglia D, Vaillancourt RE, Shepherd M, Thumma BR, Foley W, Külheim C, Potts B, Myburg AA (2012) Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet Genomes 8:463–508. doi:10.1007/s11295-012-0491-x
Hanada K, Zou C, Lehti-Shiu MD, Shinozaki K, Shiu SH (2008) Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Pl Physiol 148:993–1003. doi:10.1104/pp.108.122457
Herde M, Gärtner K, Köllner TG, Fode B, Boland W, Gershenzon J, Tholl D (2008) Identification and regulation of TPS04/GES, an Arabidopsis geranyllinalool synthase catalyzing the first step in the formation of the insect-induced volatile C16-homoterpene TMTT. Pl Cell 20:1152–1168. doi:10.1105/tpc.106.049478
Keszei A, Hassan Y, Foley WJ (2010a) A biochemical interpretation of terpene chemotypes in Melaleuca alternifolia. J Chem Ecol 36:652–661. doi:10.1007/s10886-010-9798-y
Keszei A, Webb H, Kulheim C, Foley W (2010b) Genetic tools for improving tea tree oils. Rural Industries Research and Development Corporation, Barton
Külheim C, Padovan A, Hefer C, Krause ST, Köllner TG, Myburg AA, Foley WJ (2015) The Eucalyptus terpene synthase gene family. BMC Genomics 16:1. doi:10.1186/s12864-015-1598-x
Lespinet O, Wolf YI, Koonin EV, Aravind L (2002) The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res 12:1048–1059. doi:10.1101/gr.174302
Lyons E, Freeling M (2008) How to usefully compare homologous plant genes and chromosomes as DNA sequences. Pl J 53:661–673. doi:10.1111/j.1365-313X.2007.03326.x
Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Freeling M (2008) Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Pl Physiol 148:1772–1781. doi:10.1104/pp.108.124867
Morcia C, Malnati M, Terzi V (2012) In-vitro antifungal activity of terpinen-4-ol, eugenol, carvone, 1,8-cineole (eucalyptol) and thymol against mycotoxigenic plant pathogens. Food Addit Contam A 29:415–422. doi:10.1080/19440049.2011.643458
Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Goodstein DM (2014) The genome of Eucalyptus grandis. Nature 510:356–362. doi:10.1038/nature13308
Navia-Giné WG, Yuan JS, Mauromoustakos A, Murphy JB, Chen F, Korth KL (2009) Medicago truncatula (E)-β-ocimene synthase is induced by insect herbivory with corresponding increases in emission of volatile ocimene. Pl Physiol Biochem 47:416–425. doi:10.1016/j.plaphy.2009.01.008
Penuelas J, Llusia J, Asensio D, Munné-Bosch S (2005) Linking isoprene with plant thermotolerance, antioxidants and monoterpene emissions. Pl Cell Environm 28:278–286. doi:10.1111/j.1365-3040.2004.01250.x
Pierce BA (2012) Genetics: a conceptual approach, 4th edn. WH Freeman/Macmillan, Sydney
Rambaut A (2014) Figtree: molecular evolution, phylogenetics and epidemiology. Available at: http://tree.bio.ed.ac.uk/software/figtree/. Accessed 2 May 2017
Rasoul MAA, Marei GIK, Abdelgaleil SA (2012) Evaluation of antibacterial properties and biochemical effects of monoterpenes on plant pathogenic bacteria. African J Microbiol Res 6:3667–3672. doi:10.5897/AJMR12.118
Schein AI, Kissinger JC, Ungar LH (2001) Chloroplast transit peptide prediction: a peek inside the black box. Nucl Acids Res 29:e82–e82. doi:10.1093/nar/29.16.e82
Sharkey TD, Yeh S, Wiberley AE, Falbel TG, Gong D, Fernandez DE (2005) Evolution of the isoprene biosynthetic pathway in kudzu. Pl Physiol 137:700–712. doi:10.1104/pp.104.054445
Sharkey TD, Gray DW, Pell HK, Breneman SR, Topper L (2013) Isoprene synthase genes form a monophyletic clade of acyclic terpene synthases in the tps-b terpene synthase family. Evolution 67:1026–1040. doi:10.1111/evo.12013
Shelton D, Aitken K, Doimo L, Leach D, Baverstock P, Henry R (2002) Genetic control of monoterpene composition in the essential oil of Melaleuca alternifolia (Cheel). Theor Appl Genet 105:377–383. doi:10.1007/s00122-002-0948-7
Shelton D, Leach D, Henry R (2004a) Isopentenyl pyrophosphate isomerases from Melaleuca alternifolia (Cheel) and their role in isoprenoid biosynthesis. J Hortic Sci Biotech 79:289–292. doi:10.1080/14620316.2004.11511762
Shelton D, Zabaras D, Chohan S, Wyllie SG, Baverstock P, Leach D, Henry R (2004b) Isolation and partial characterisation of a putative monoterpene synthase from Melaleuca alternifolia. Pl Physiol Biochem 42:875–882. doi:10.1016/j.plaphy.2004.10.010
Shepherd M, Ablett G, Wood R, Raymond C, Rose T (2015) Ecotype variation in early growth, coppicing, and shoot architecture of tea tree (Melaleuca alternifolia). Industr Crop Prod 76:844–856. doi:10.1016/j.indcrop.2015.07.076
Shimoda T, Nishihara M, Ozawa R, Takabayashi J, Arimura GI (2012) The effect of genetically enriched (E)-β-ocimene and the role of floral scent in the attraction of the predatory mite Phytoseiulus persimilis to spider mite-induced volatile blends of torenia. New Phytol 193:1009–1021. doi:10.1111/j.1469-8137.2011.04018.x
Silva SM, Abe SY, Murakami FS, Frensch G, Marques FA, Nakashima T (2011) Essential oils from different plant parts of Eucalyptus cinerea F. Muell. ex Benth.(Myrtaceae) as a source of 1, 8-cineole and their bioactivities. Pharmaceuticals 4:1535–1550. doi:10.3390/ph4121535
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Soltis P, Soltis D (2003) Applying the bootstrap in phylogeny reconstruction. Statist Sci 18:256–267. doi:10.1214/ss/1063994980
Steele CL, Crock J, Bohlmann J, Croteau R (1998) Sesquiterpene synthases from grand fir (Abies grandis) comparison of constitutive and wound-induced activities, and cDNA isolation, characterization, and bacterial expression of δ-selinene synthase and γ-humulene synthase. J Biol Chem 273:2078–2089. doi:10.1074/jbc.273.4.2078
Tao N, Jia L, Zhou H (2014) Anti-fungal activity of Citrus reticulata Blanco essential oil against Penicillium italicum and Penicillium digitatum. Food Chem 153:265–271. doi:10.1016/j.foodchem.2013.12.070
Thornhill AH, Ho SY, Külheim C, Crisp MD (2015) Interpreting the modern distribution of Myrtaceae using a dated molecular phylogeny. Molec Phylogen Evol 93:29–43. doi:10.1016/j.ympev.2015.07.007
Toyomasu T, Tsukahara M, Kaneko A, Niida R, Mitsuhashi W, Dairi T, Sassa T (2007) Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc Natl Acad Sci USA 104:3084–3088. doi:10.1073/pnas.0608426104
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Schein J (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604. doi:10.1126/science.1128691
Webb H, Lanfear R, Hamill J, Foley WJ, Külheim C (2013) The yield of essential oils in Melaleuca alternifolia (Myrtaceae) is regulated through transcript abundance of genes in the MEP pathway. PLoS ONE 8:e60631. doi:10.1371/journal.pone.0060631
Webb H, Foley WJ, Külheim C (2014) The genetic basis of foliar terpene yield: implications for breeding and profitability of Australian essential oil crops. Pl Biotechnol 31:363–376. doi:10.5511/plantbiotechnology.14.1009a
Wilson LJ, Bauer LR, Walter GH (1996) ‘Phytophagous’ thrips are facultative predators of twospotted spider mites (Acari: Tetranychidae) on cotton in Australia. Bull Entomol Res 86:297–305. doi:10.1017/S0007485300052597
Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE, Ikeda H (2015) Terpene synthases are widely distributed in bacteria. Proc Natl Acad Sci USA 112:857–862. doi:10.1073/pnas.1422108112
Yamaguchi S (2008) Gibberellin metabolism and its regulation. Annual Rev Pl Biol 59:225–251. doi:10.1146/annurev.arplant.59.032607.092804
Acknowledgements
The authors wish to acknowledge the assistance of R. Wood, A. Kawamata and J. Bloomfield and T. Rhodes for his help in the laboratory. Jed Calvert would also like to thank Shirali, for her constant support and supply of fresh perspectives. This work was supported by the Australian Research Council (Grant No. DP140102552).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling editor: Yunpeng Zhao.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1: Methods and results for generating a draft genome sequence for Melaleuca alternifolia.
Online Resource 2: Flow cytometry methods for genome size estimation in Melaleuca alternifolia.
Online Resource 3: TPS subfamily conserved amino acid sequences used as queries for BLASTing the Melaleuca alternifolia assembly. Originally published in a supplementary file with Külheim et al. 2015.
Online Resource 4: tBLASTn searches of Melaleuca alternifolia (Southern Cross Plant Science unmasked vv1.1) genome using queries listed in Kulheim et al. (2015) (Calvert et al._ESM_3.pdf) to find TPS genes not predicted by MAKER v2.31.8.
Online Resource 5: Top e-values for conserved TPS subfamily domain queries in Melaleuca alternifolia and Eucalyptus grandis.
Online Resource 6: Maximum-likelihood tree produced using the 113 amino acid sequences of Eucalyptus grandis TPS genes (from subfamilies a, b, c, e, f and g) identified by Külheim et al. (2015). Tree is rooted at the branching of type I and III genes. Phylogeny shows high structural similarity to Külheim et al. down to branch length, which denotes relationship distance. Scale = average number of amino acid substitutions per branch (JPEG produced using Figtree v1.4.2. And GIMP).
Online Resource 7: FASTA Alignment of 113 E. grandis TPS genes plus the 37 Melaleuca alternifolia candidate gene models identified using BLAST, as well as the coding sequence for a putative monoterpene synthase transcript obtained by Shelton et al. (2004a, b; GenBank accession AY279379.1). Using PhyML 3.0 (http://phylogeny.lirmm.fr; Dereeper et al. 2008) with default settings, a ClustalW alignment was constructed from the 113 sequences. Gblocks curation was skipped.
Online Resource 8: 37 Melaleuca alternifolia TPS gene models listed by subfamily. Quality class ranking as per Külheim et al. (2015) is as follows: 1 = Full length, no prem stop codons; 2 = Full length, up to 2 stop codons; 3 = Full length, no stop codon; 4 = Pseudogenes, more than 2 stop codons; 5 = Partial gene.
Online Resource 9: Amino acid sequences of thirty-seven candidate TPS genes with high similarity to conserved TPS regions identified in the Melaleuca alternifolia genome.
Online Resource 10: Melaleuca alternifolia and Eucalyptus grandis gene models predicted by ChloroP 1.1 to contain chloroplast transit peptides.
Online Resource 11: Melaleuca alternifolia TPS gene models (37) analysed using PCLR (Schein et al. 2001) for the presence of chloroplast transit peptides (cTP).
Online Resource 12: Phylogenetic tree file. Replication of the Külheim et al. 2015 phylogenetic tree for TPS genes in Eucalyptus grandis, used as a foundation for comparative analysis.
Online Resource 13: Phylogenetic tree file. Replication of the Külheim et al. 2015 phylogenetic tree for TPS genes in Eucalyptus grandis, with inclusion of 37 Melaleuca alternifolia putative TPS genes and one putative monoterpene synthase gene (Shelton et al. 2004a, b).
Online Resource 14: Methods for collection of C. citriodora subsp. variegata data provided in Table 1.
Rights and permissions
About this article

Cite this article
Calvert, J., Baten, A., Butler, J. et al. Terpene synthase genes in Melaleuca alternifolia: comparative analysis of lineage-specific subfamily variation within Myrtaceae. Plant Syst Evol 304, 111–121 (2018). https://doi.org/10.1007/s00606-017-1454-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-017-1454-3