
Abstract
Amlodipine (AM) is a long active calcium channel blocker used to relax blood vessels by preventing calcium ion transport into the vascular walls and its supporting molecules acetaminophen (AP) and ascorbic acid (AA) are recommended for hypertension control and prevention. Considering their therapeutic importance and potential side effects due to over dosage, we have fabricated a sensor for individual and simultaneous determination of AA, AP, and AM in pharmaceuticals and human urine using novel Zn-doped Ca2CuO3 nanoparticles modified glassy carbon electrode (GCE). Optimally doped Ca2CuO3 (2.5 wt% Zn at Cu site) enhanced the detection of target molecules over much wider concentration ranges of 50 to 3130 µM for AA, 0.25 to 417 µM for AP, and 0.8 to 354 µM for AM with the corresponding lowest detection limits of 14 µM, 0.05 µM, and 0.07 µM, respectively. Furthermore, the Zn-Ca2CuO3/GCE exhibited excellent selectivity and high sensitivity even in the presence of several potential interfering agents. The usefulness of the developed electrode was tested using an amlodipine besylate tablet and urine samples of seven hypertension patients under medication. The results confirmed the presence of a significant amount of AP and AM in six patients’ urine samples indicating that the personalized medication is essential and the quantum of medication need to be fixed by knowing the excess medicines excreted through urine. Thus, the Zn-Ca2CuO3/GCE with a high recovery percentage and good sensitivity shall be useful in the pharmaceutical and biomedical sectors.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Drug overdose can result in serious health complications and even death. In particular, the number of causalities caused by excessive usage of cardiovascular drugs continues to increase each year. Cardiovascular drugs are used to lower blood pressure in patients and as vasodilators in blood vessels to increase blood flow [1]. Amlodipine (AM), C20H25ClN2O5, a long-acting drug with a half-life of 30–58 h [2], is the most frequently recommended medication for treating hypertension patients. In contrast to other hypertension medications, it is administered as a single dosage per day. AM is a third-generation calcium channel blocker, and is a substituted dialkyl 1,4-dihydropyridine-3,5-dicarboxylate derivative [3]. It is used to treat cardiac arrhythmias, angina pectoris, and vasospastic angina as well as high blood pressure and hypertension [4, 5]. It works by limiting the entry of calcium ions into cardiac and vascular smooth muscles, protecting the target organs [6]. Consuming excessive amounts of AM leads to serious health issues like palpitations, nausea, leg swelling, and fatigue [7, 8]. Furthermore, at higher dosage levels, it causes hypotension [9] and shock [10].
Acetaminophen or paracetamol is a commonly utilized antipyretic and analgesic medication for alleviating mild to moderate pains such as headaches, toothaches, muscle aches, and backaches [11, 12]. Additionally, it is employed to lower fevers associated with viral and bacterial infections [13]. While acetaminophen generally lacks noticeable side effects, prolonged and excessive consumption may result in significant hepatotoxicity and nephrotoxicity [14]. Elevated levels of acetaminophen have been linked to the development of asthma and eczema in children under 1 year of age. Sometimes, it is recommended to take AP along with AM. Ascorbic acid (AA), also known as vitamin C, is recognized for its potent antioxidant properties against various free radicals like hydroxyl and superoxide [15]. It finds applications as an antioxidant in food, animal feed, and cosmetics [16]. Understanding the concentration of AA in biological fluids is crucial for assessing oxidative stress levels. AA has been extensively employed in treating conditions such as colds, scurvy, and mental illnesses [17]. Generally, zinc tablet was prescribed with vitamin C tablets to boost the immunity, and zinc and vitamin C help the human body to act against numerous ailments. Hence, simultaneous determination of AM and AP in addition with AA is of great clinical importance. As a result, it is necessary to develop a sensitive and simple detection tool for the simultaneous determination of AA, AP, and AM in the pharmaceutical and biomedical fields.
Electrochemical sensors have been developed to determine AM in pharmaceutical tablets and clinical samples because of their advantage over the traditional methods (HPLC, GC-MS, electrophoresis, and spectrophotometry). Sudha et al. reported a silver-cerium tungstate-decorated carbon nanofiber composite-based electrochemical sensor platform for the detection of AM in pharmaceutical tablets and human urine [5]. In another study, a NiMoO4/chitosan nanocomposite-based voltammetric sensor was reported for AM detection [8]. The use of a CuO-NiO nanocomposite and an ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate-modified carbon paste electrode to determine AM in the linear concentration range of 0.1 to 100 µM has been reported [3]. Khairy et al. used DNA to modify a screen-printed electrode (SPE) and employed it to diagnose AM [11] in tablet and human urine. Atta et al. have used multi-walled carbon nanotubes (CNT), ionic liquid crystal (ILC), graphene oxide (RGO), and crown ether (CW) (18-Crown-6) to modify the glassy carbon electrode which yielded LODs for AA, AP, and AM of 9.24 nM, 0.0906 nM, and 0.139 nM, respectively [18]. Veerapandi et al. have synthesized Ca2CuO3 by co-precipitation method and employed it for the determination of xanthine derivatives [19]. Lavanaya et al. have reported that co-doping into CeO2 induced the ferromagnetic behaviour in the parent compound CeO2 and this chemically modified material was used as electrode material for the detection of xanthine and its analogues [20].
Nanostructured metal oxides have been widely used for sensor fabrication because of their tunable physical, chemical properties, and large-scale availability at low cost [14]. Here we report Ca2CuO3, a new electrode material with high electrocatalytic activity towards the detection of AA, AP, and AM at well-separated potentials. Ca2CuO3 is a 1D perovskite material in the family of high temperature superconductor with antiferromagnetic behaviour at a Neel temperature (TN) of ~8 K [21]. Zn doping into Ca2CuO3 system enhanced the electrochemical characteristics towards the oxidation of AA, AP, and AM. The modified electrode exhibited high stability and sensitivity without any other mediator or promotor.
Materials and methods
Chemicals
The chemicals used in the present work were purchased from different companies as listed below along with their purity: copper nitrate (98%, Alfa Aesar, England), calcium nitrate (98%, Nice Chemicals Pvt. Ltd., Kochi), sodium hydroxide (98%, Fisher Scientific, Mumbai), potassium hexacyanoferrate (III), potassium hexacyanoferrate (II) and disodium hydrogen phosphate (99%, Merck specialties Pvt. Ltd., Mumbai), potassium chloride (99%, Loba Chemie Pvt. Ltd., Mumbai), sodium dihydrogen phosphate (99%, Spectrochem, Mumbai), and ascorbic acid, amlodipine besylate, and acetaminophen (98%, Sisco Research Laboratory, Mumbai).
Synthesis of pure and Zn-doped Ca2CuO3
Ca2CuO3 nanoparticles was synthesized by chemical co-precipitation method as described in our previous report [19]. In order to prepare pristine Ca2CuO3 powders, appropriate amounts of Ca(NO3)2 (5.904 g) and Cu(NO3)2 (2.907 g) were weighed and the individual solutions were prepared separately using deionized water and mixed thoroughly using a magnetic stirrer running at 400 rpm. 2 M NaOH was added dropwise to this metal nitrate solution while stirring until the pH of the solution reached 12–13 and the obtained precipitates were washed several times with deionized water and then with ethanol. The resultant black colour powder was dried at 80 °C and then annealed at 600 °C for 3 h to obtain the final product. For synthesizing Ca2(Cu1 − xZnx)O3 (x = 2.5 wt% and 5 wt%), appropriate amounts of Zn(NO3)2 (0.372 g for 2.5 wt% and 0.744 g for 5 wt%) have been added to 5.904 g of Ca(NO3)2 and Cu(NO3)2 (2.617 g for 2.5 wt% and 2.326 g for 5 wt%) and the above procedure was followed. The 2.5 wt% Zn-doped and 5 wt% Zn-doped Ca2CuO3 were named as 2.5Zn-Ca2CuO3 and 5Zn-Ca2CuO3.
Instruments
Powder X-ray diffraction (XRD) measurement was performed using X'Pert Pro PANalytical to analyze the crystal structure of the synthesized nanoparticles. High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) were performed using the JEOL JEM 2100 microscope. Magnetic studies have been performed using Vibrating Sample Magnetometer (VSM) (Cryogenic).
Electrochemical measurements were conducted using the potentiostat PGSTAT 204 (Autolab, Metrohm). A conventional three-electrode system was employed, consisting of a glassy carbon electrode (~3 mm dia) coated with Ca2CuO3 and Zn-Ca2CuO3 nanoparticles serving as the working electrode, an Ag/AgCl electrode (3 M KCl) utilized as the reference electrode, and a platinum wire serving as the counter electrode. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were carried out in a 0.1 M KCl solution containing a 1 mM [Fe(CN)6]3−/4− redox couple for basic electrochemical studies. EIS measurements were carried out across a frequency range spanning from 100 kHz to 1 Hz, with a DC potential of 250 mV and an AC potential of ± 5 mV.
Furthermore, the sensing experiments were conducted in PBS (pH 7.0) using both CV and square wave voltammetry (SWV) techniques. SWV experiments were conducted with a frequency set at 15 Hz, amplitude of 0.025 V, and an increment of 0.004 V.
Fabrication of Zn-Ca2CuO3 nanoparticles modified GCEs
Initially, surfaces of GCE (~3 mm dia) were renewed by cleaning it with 0.05 µ alumina powder and then sonicating it in ethanol and water mixture for about 10 min. The electrode was then activated by performing ten continuous cyclic voltammograms (CVs) in 0.1 M sulphuric acid at 0.05 V/s between potentials of 0.1 to 1.0 V using Ag/AgCl as reference electrode. Then, 1.0 mg of Ca2CuO3, 2.5Zn-Ca2CuO3, and 5Zn-Ca2CuO3 were weighed separately, and 1.0 mL deionized water was added to each sample separately. They are then ultrasonically treated for about 10 min to obtain a uniform solution. Ten microliters of this suspension was drop casted on the mirror polished GCE surface and allowed to dry at room temperature. The prepared electrodes were named as Ca2CuO3/GCE, 2.5Zn-Ca2CuO3/GCE, and 5Zn-Ca2CuO3/GCE. Ag/AgCl and Pt wire were employed as reference and counter electrode, respectively.
Preparation of tablet solution
Amlodipine besylate tablet (Amlip-5) purchased from a local medical store contained 5 mg of amlodipine as well as starch, sodium carbonate, and other excipients. Using a mortar and pestle, five tablets amounting 100 mg in mean weight were crushed into a fine powder, dissolved in a 50 mL standard flask with an ethanol and water mixture and the solution was centrifuged and filtered. Different volumes (250 to 1000 µL) of this supernatant solution were added into the electrochemical cell.
Preparation of urine samples
Urine samples were collected from seven patients under medications. The samples were centrifuged at 6000 rpm for 15 min, filtered through Whatman filter paper (Grade 1, pore size 11 μm), and used for analyses without any other pretreatment.
Results and discussion
Physical characterizations
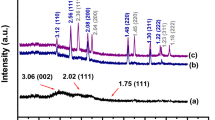
Figure 1A depicts the powder X-ray diffraction patterns of synthesized pure Ca2CuO3, 2.5Zn-Ca2CuO3, and 5Zn-Ca2CuO3 nanomaterials annealed at 600 °C for 3 h in ambient atmosphere. All of the obtained diffraction peaks match well with the reported values [19] and ICDD pattern no. 01-072-0781. The XRD patterns obtained for the Zn doped materials show that all of the diffraction peaks corresponded to pure Ca2CuO3 and no other diffraction peaks corresponding to possible impurities such as CaO, CuO, and ZnO or any other phases were observed. It was noted that the diffraction peak intensities increased while the peak width (FWHM) of strong peaks (301) and (011) decreased with Zn ion doping. These changes in the diffraction pattern are due to doping of Zn (74 pm) into Cu (73 pm). The average crystallite sizes for Ca2CuO3, 2.5Zn-Ca2CuO3, and 5Zn-Ca2CuO3 nanomaterials were calculated (using Debye Scherrer formula) to be 15.2 nm, 23.5 nm, and 22.1 nm, respectively, and the corresponding dislocation density strain values was given in Table ST1. The obtained values indicated that the incorporation of Zn (II) ions into Cu (II) ions has resulted in difference in the values of dislocation density and micro stain.

A P-Xrd pattern of (a) Ca2CuO3, (b) 2.5Zn-Ca2CuO3, and (c) 5Zn- Ca2CuO3; B wide scan XPS spectra of 2.5Zn-Ca2CuO3; deconvoluted XPS spectra of (C) Ca 2p, (D) Cu 2p, (E) O 1s, and (F) Zn 2p
X-ray photoelectron spectrum (XPS) was recorded for 2.5Zn-Ca2CuO3 NPs to confirm the chemical states of the elements. The wide scan XPS spectrum (Fig. 1B) shows peaks at 347.2 eV and 350.4 eV corresponding to Ca2p, 933.8 eV and 953.7 eV for Cu2p, 530.9 eV for O1s, and 1022.3 eV and 1045.3 eV for Zn 2p, respectively. The deconvoluted Ca 2p spectra showed two peaks at the binding energies of 347.2 eV for Ca 2p3/2 and 350.4 eV for Ca 2p1/2 (Fig. 1C). The difference of 3.2 eV (spin-orbit splitting value) between these peaks indicated that the valency of Ca is + 2. Figure 1D shows the deconvoluted Cu 2p spectra exhibiting two major peaks at the binding energies of 933.8 eV and 953.7 eV corresponding to Cu 2p3/2 and Cu 2p1/2. Also, two strong satellite peaks pertaining to Cu (II) ion were observed at 940.2 eV and 942.7 eV. The spin-orbit splitting value of 19.9 eV confirmed the presence of Cu in the + 2-oxidation state. The O1s spectra shows a peak at 530.9 eV corresponding to O2− ions (Fig. 1E). Figure 1F shows the deconvoluted Zn2p spectra with two peaks at the binding energies of 1022.2 eV (Zn 2p3/2) and 1045.3 eV (Zn 2p1/2) and the difference between these two peaks (23.1 eV) confirmed the occurrence of Zn with + 2 oxidation state. Thus, the XPS results confirmed the presence of Ca, Cu, O, and Zn in the sample and their oxidation states.
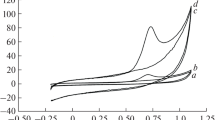
The magnetic properties of Ca2CuO3, 2.5Zn-Ca2CuO3, and 5Zn-Ca2CuO3 samples were measured at room temperature to figure out the impact of magnetic properties on the electrocatalytic behaviour (Fig. 2A). The Ca2CuO3 displays a negative M-H curve, and the absence of a hysteresis loop demonstrated its diamagnetic nature (Ms = −0.85 × 10−3 emu/g). The 2.5% Zn-doped sample exhibited a hysteresis like loop with negative susceptibility, and when the dopant quantity was increased to 5%, the susceptibility curve was found to retain the magnetic curve of pure compound. This result showed that the magnetic nature of Ca2CuO3 was changed by the addition of Zn, and the saturation magnetization values (Ms) for the 2.5Zn-Ca2CuO3 and 5Zn-Ca2CuO3 samples were determined to be 0.07 × 10−3 emu/g and − 0.31 × 10−3 emu/g, respectively. The Ms value for 2.5Zn-Ca2CuO3 was found to be high, demonstrating that 2.5% doping has a high order of magnetism than 5Zn-Ca2CuO3. The oxygen vacancies generated by the doping of Zn may be the cause of the change in magnetic behaviour.

A Magnetic susceptibility curves of (a) Ca2CuO3, (b) 2.5Zn-Ca2CuO3, and (c) 5Zn-Ca2CuO3; B–C TEM, D HRTEM, and E SAED pattern of 2.5Zn-Ca2CuO3
Figure 2B–D shows the TEM and HRTEM images of 2.5Zn-Ca2CuO3 sample. The TEM images clearly evidenced the spherical morphology with variant sizes. The size of the one sphere shown in Fig. 2C was found to be 70 nm (ImageJ software was used to calculate the size). The d-spacing value was calculated from the HRTEM image recorded at 10-nm magnification. The calculated d-spacing value of 205 nm corresponding to (301) plane of the XRD pattern confirms the good agreement between the d-spacing values of XRD and HRTEM results. The selected area diffraction pattern of 2.5Zn-Ca2CuO3 sample shows the bright spotty rings and the spacing between the rings have been calculated using the ImageJ software. The values of 205 nm and 210 nm were in good agreement with the XRD and HRTEM results.
Electrochemical characterization of nanoparticles modified GCEs
Figure S1A shows the cyclic voltammograms recorded at (a) bare GCE, (b) Ca2CuO3/GCE, (c) 2.5Zn-Ca2CuO3/GCE, and (d) 5Zn-Ca2CuO3/GCE in 1 mM [Fe(CN)6]3−/4− containing 0.1 M KCl at a scan rate of 0.05 V/s. The nanomaterials modified electrodes demonstrated remarkable fast electron transfer ability for the redox reaction of [Fe(CN)6]3−/4− when compared to bare GCE. Values of 16.6 µA, 18.2 µA, 20.1 µA, and 19.02 µA for bare GCE, Ca2CuO3/GCE, 2.5Zn-Ca2CuO3/GCE, and 5Zn-Ca2CuO3/GCE, respectively, have been obtained. When compared to other electrodes, the 2.5Zn-Ca2CuO3/GCE has relatively higher peak current and the electron transport ability of Zn-doped samples is superior to that of undoped and unmodified GCEs. In addition, 2.5Zn-Ca2CuO3/GCE showed the peak separation value of 80 mV which is lower than that of bare GCE (116 mV), Ca2CuO3/GCE (94 mV), and 5Zn-Ca2CuO3/GCE (91 mV). The observed results suggest that the optimum Zn-loading is 2.5% at Cu-site which results in smaller Ca2CuO3 with higher surface area for the reactions to occur and high electron transfer rate. Based on these results, 2.5Zn-Ca2CuO3/GCE was chosen for further investigation.
CVs recorded at 2.5Zn-Ca2CuO3/GCE as a function of scan rate (0.01 V/s to 0.1 V/s) in 1 mM [Fe(CN)6]3−/4− containing 0.1 M KCl are shown in Fig. S1B. The redox peak currents were found to increase with scan rate, indicating a diffusion-controlled electrode process. The plot of the square root of the scan rate versus the redox peak current revealed a linear relationship with the regression equation Ipa = 81.95ν1/2 (V1/2 s−1/2) − 0.082 (R2 = 0.9989) and Ipc = −49.48ν1/2 (V1/2 s−1/2) – 1.99 (R2 = 0.9938) which confirms the diffusion-controlled electrode process. Further, the relationship between square root of scan rate and peak current was given by Randles-Sevcik equation:
The electrochemical active surface area of the electrodes was calculated using the Eq. (1) as 0.088 cm2, 0.096 cm2, 0.115 cm2, and 0.105 cm2 for bare GCE, Ca2CuO3/GCE, 2.5Zn-Ca2CuO3/GCE, and 5Zn-Ca2CuO3/GCE. The 2.5Zn-Ca2CuO3/GCE has a large electroactive surface area for the reaction to take place on the electrode.
Electrochemical impedance spectroscopy was performed in 1 mM [Fe(CN)6]3−/4− containing 0.1 M KCl in the frequency region from 100 kHz to 1 Hz at a DC potential of 250 mV and an AC potential of 5 mV to investigate the electrode-electrolyte interface property. The Nyquist plots for (a) bare GCE, (b) Ca2CuO3/GCE, (c) 2.5Zn-Ca2CuO3/GCE, and (d) 5Zn- Ca2CuO3/GCE are shown in Fig. S1C. The Nyquist plots were fitted using Randles equivalent circuit model (inset Fig. S1C) and the charge transfer resistance values have been calculated. The Rct value of 2.5Zn-Ca2CuO3/GCE (2485 ohm/cm2) is lower than that of bare GCE (7204 ohm/cm2), Ca2CuO3/GCE (5241 ohm/cm2), and 5Zn-Ca2CuO3 (4820 ohm/cm2). The reason for the lower Rct in 2.5Zn-Ca2CuO3-modified glassy carbon electrode could be due to optimal hole doping at this doping level though replacement of Cu2+/1+ by Zn2+ and that the higher doping concentration leads to particle agglomeration and subsequent reduction in surface area. This inference is corroborated by cyclic voltammetry experiments, which revealed a diminished active surface area for the 5Zn-Ca2CuO3/GCE when compared to that of 2.5Zn-Ca2CuO3. This reduction in the surface area ultimately hinders the electron transfer rate through the electrode-electrolyte surface. Consequently, the 5Zn-Ca2CuO3/GCE demonstrated a higher Rct compared to the 2.5Zn-Ca2CuO3/GCE. The lower Rct value indicated that the 2.5Zn-Ca2CuO3/GCE has higher electron transfer kinetics than the other electrodes.
Effect of pH on the oxidation reactions at Zn-Ca2CuO3/GCE
The pH of the supporting medium influences the redox behaviour of all analytes. To find out the optimum value, PBS with pH ranging from 3 to 10 was prepared and its effect on the oxidation reactions of AA, AP, and AM at the 2.5Zn-Ca2CuO3/GCE was investigated. Square wave voltammograms (SWVs) recorded in the presence of 10 µM of each AA, AP, and AM at the modified electrode in PBS of different pH are shown in Fig. 3A. At a lower pH of 3.0, the target analytes were oxidized at a higher oxidation potential of 0.42 V, 0.68 V, and 0.88 V for AA, AP, and AM, respectively. As the pH rises, the oxidation peak shifts to lower potentials. This clearly demonstrated the involvement of protons (H+) in the oxidation process of AA, AP, and AM at the electrode surface. A plot of pH versus anodic peak potentials (Epa) of each analyte clearly shows a linear relationship with an R2 of 0.983, 0.95, and 0.961, respectively, for AA, AP, and AM. This plot yields the regression equation Ep (V) = 1.005–0.042pH, Ep (V) = 1.243–0.048pH, and Ep (V) = 1.401–0.047pH. The slope values of 0.042, 0.048, and 0.047 were found to nearly close with the theoretical slope value of 0.059 V which indicates that the oxidations of AA, AP, and AM at the modified electrode surface involves an equal number of protons and electrons [19, 22]. Further, the number of electrons transferred during the oxidation reactions have been calculated using the following formula [2]:

A SWVs recorded in the presence of 500 µM AA, 5 µM each AP and AM to study the effect of pH (3.0 to 10.0) in 0.1 M PBS. B CVs and D SWVs obtained in 0.1 M PBS (pH 7.0) containing 500 µM AA, 5 µM each AP and AM at (a) bare GCE, (b) Ca2CuO3/GCE, (c) 2.5Zn-Ca2CuO3/GCE and 5Zn-Ca2CuO3/GCE. C CVs of 2.5Zn-Ca2CuO3/GCE shown as a function of scan rate (0.05–0.1 V/s)
where m is the number of electrons transferred, n is the number of protons transferred, and the other constants have their usual meanings. The number of electrons transferred was calculated as 2 for all the three analytes (AA, AP, and AM).
Further, the peak currents of AA and AM increases from pH of 3.0 to the maximum at pH of 7.0. On the other hand, AP showed higher peak current value at pH 5. However, considering better peak-to-peak separation value (102 mV for AA and AP and 300 mV for AP and AM) and higher anodic currents for AA and AM, the pH of 7.0 was chosen for further investigations.
A proposed reaction mechanism for the detection of AA, AP, and AM at the electrode surface is as follows: The oxidation of AA at the electrode surface results in the removal of two protons from the hydroxyl (-OH) groups located at the 3rd and 4th positions of the furan ring, accompanied by the loss of two electrons, yielding dehydroascorbic acid [23]. For AP, oxidation occurs at the amide (-NH) group, leading to its conversion into an amine (= N-) moiety. This process is followed by the detachment of two protons and two electrons [24]. AM undergoes oxidation, where the pyridine (-NH-) group is transformed into a = N- group, with the simultaneous loss of two protons and two electrons [5] (Supplementary Scheme 1).
Electrochemical behaviour of AM, AP, and AP at 2.5Zn-Ca2CuO3/GCE
Cyclic voltammograms were measured in 0.1 M PBS (pH 7.0) containing 10 µM of each AA, AP and AM at (a) bare GCE, (b) Ca2CuO3/GCE, (c) 2.5Zn-Ca2CuO3/GCE, and (d) 5Zn-Ca2CuO3/GCE in the potential range of 0 to 1.0 V at a step potential of 0.05 V/s (Fig. 3B). In PBS, none of the electrodes did respond, whereas in the presence of analytes, all of the electrodes exhibited peaks corresponding to the oxidation of AA, AP, and AM. The absence of a reduction peak confirmed that AA, AP, and AM were irreversibly electro-oxidized at the electrode surface. Among the investigated electrodes, 2.5Zn-Ca2CuO3/GCE demonstrated a more clearly defined oxidation peak with a higher current than the other electrodes. Moreover, the 2.5Zn-Ca2CuO3/GCE electrode oxidized the analytes at the lower potentials than the other electrodes and higher peak current values of 3.21 µA, 4.06 µA, and 3.73 µA for AA, AP, and AM, respectively.
The effect of scan rate on the oxidation behaviour of AA, AP, and AM was investigated between step potentials of 0.02 to 0.1 V/s with a 0.01 V/s increment at each step. The CVs recorded in 0.1 M PBS (pH 7.0) at 2.5Zn-Ca2CuO3/GCE are shown in Fig. 3C. The oxidation peak was found to increase with each step potential increment, with a shift towards higher potentials, confirming the irreversible electrode reaction at the electrode surface. It implies that the diffusion process controls the oxidation reaction of AA, AP, and AM at the electrode surface.
Figure 3D shows the square wave voltammogram of 0.1 M PBS (pH 7.0) containing 10 µM of each AA, AP, and AM recorded in the potential range of 0.1 to 1.0 V (Fig. 4C). The oxidation peak currents of AA, AP, and AM were found to be much higher at 2.5Zn-Ca2CuO3/GCE than at the other electrodes, supporting the findings from CV studies. It is noticed from the voltammograms that the bare GCE suffers to oxidize AA and AP at different potentials. Also, Ca2CuO3/GCE showed the small response for the oxidation of AA. On the other hand, the electrodes modified with 2.5Zn- and 5Zn-doped Ca2CuO3 exhibited well-resolved oxidation peaks for AA and AP at distinct potentials with high current. This remarkable electrocatalytic activity towards the oxidation at different potential may be due to affinity of AA to Zn. Ascorbic acid has the tendency to form co-ordination complex with the Zn ions, as a result of this interaction, the electrode modified with Zn-doped Ca2CuO3 possesses affinity for AA. Thereby, it is proposed that the ability of the Zn-Ca2CuO3-modified electrode for the detection of analytes at different potential is due to the doping of Zn into Ca2CuO3. In addition, optimal doping of Zn ion into Cu ion of the Ca2CuO3 system provides a high reactive surface area and speeds up the charge transfer rate compared to the other electrodes. These results demonstrated clearly that the 2.5Zn-Ca2CuO3/GCE electrode is suitable for determining AA, AP, and AM simultaneously.

A SWVs obtained for the various concentrations of AA (50 to 3130 µM); B the plot of anodic peak current against the concentration of AA. C SWVs obtained for the various concentrations of AP (0.25 to 417 µM); D the plot of anodic peak current against the concentration of AP. E SWVs obtained for the various concentrations of AM (0.3 to 354 µM); F the plot of anodic peak current against the concentration of AM
Selective and simultaneous determinations of AM, AP, and AP
The square wave voltammograms were recorded at 2.5Zn-Ca2CuO3/GCE in 0.1 M PBS (pH 7.0) for various concentrations of AA (50 to 3130 µM) containing a fixed amount of AP and AM (25 µM of each) and the results are shown in Fig. 4A. As can be seen, the voltammetric responses increased as the concentration of AA increased. The plot of concentration of AA versus anodic current (Fig. 4B) demonstrates the presence of a linear relationship with the following regression equations:
-
Ip (µA) = 0.0003CAA + 0.043 (R2 = 0.998).
Similarly, the concentration of AP was varied while maintaining other two analytes at constant value (Fig. 4C–D). The electrode exhibited two linear range over the concentration ranges of 0.25 µM to 85.5 µM and 85.5 µM to 417 µM with following regression equations:
-
Ip (µA) = 0.0048CAP + 0.016 (R2 = 0.995) (0.8 to 18 µM).
-
Ip (µA) = 0.0021CAP + 0.221 (R2 = 0.996) (18 to 354 µM).
Also, the electrode showed linear range of 0.8 to 18 µM and 18 to 354 µM while increasing the concentration of AM at the electrode (Fig. 4E–F). The corresponding linear segments with the regression equations are as follows:
-
Ip (µA) = 0.0114CAM + 0.0069 (R2 = 0.994) (0.25 to 85.5 µM).
-
Ip (µA) = 0.0036CAM + 0.135 (R2 = 0.999) (85.5 to 417 µM).
The limit of detection (LOD) for AM, AP, and AA at a S/N ratio of 3 was estimated to be 14 µM, 0.05 µM, and 0.07 µM.
Figure 5A shows the SWVs obtained at the 2.5Zn-Ca2CuO3/GCE for the simultaneous addition of AA, AP, and AM over a concentration range of 50 to 376 µM for AA and 0.5 to 18.8 µM for AP and AM. It was found that all the voltammetric responses increased linearly with increasing concentration of AA, AP, and AM. It was also observed that there were no shifts or changes among the obtained voltammograms, which indicates the suitability of the electrode for the determination of AA, AP, and AM simultaneously at the 2.5Zn-Ca2CuO3-modified GCE. The linear regression equations obtained (Fig. 5B–D) are as follows:
-
Ip (µA) = 0.0012CAA − 0.067 (R2 = 0.998) (for AA).
-
Ip (µA) = 0.0367CAP − 0.1176 (R2 = 0.995) (for AP).
-
Ip (µA) = 0.0154CAM − 0.027 (R2 = 0.992) (for AM).

A SWVs obtained for the simultaneous addition of AA (50 to 376 µM), AM (0.5 to 18.8 µM), and AP (0.5 to 18.8 µM) at 2.5Zn-Ca2CuO3/GCE in phosphate buffer (pH 7.0). B, C, D Anodic peak current against the concentration
The LODs for the simultaneous determination are deduced to be 20 µM for AA, 0.14 µM for AP, and 0.11 µM for AM. The obtained results were compared to previously reported literature results (Table 1). It was noted that the electrode developed in this study had a wider linear range and a lower LODs than the Pt-NiO/MWCNTs/GCE [25], TU/Au/CNT [26], GLNFCNTCP-SDS [4], GC/CNT/ILC/RGO/CW [18], and SEP/MWCNTs/pPG [27]. Most of these authors have used the mediators such as CNTs, ionic liquids, and metal nanoparticles to improve the sensing characteristics of the electrode. In particular, the electrode GC/CNT/ILC/RGO/CW [18] is made up of complicated structure to determine AA, AP, and AM along with dobutamine simultaneously. On the other hand, the 2.5Zn-Ca2CuO3 as single electrode material without any mediator or binder successfully oxidized AA, AP, and AM at distinct oxidation potentials without interfering each other over wider linear ranges. The fabrication process for 2.5Zn-Ca2CuO3/GCE is simpler and less expensive than that of the other electrodes reported. As a result, we assure that the electrode developed in this study could be the best electrode for determining AA, AP, and AM in pharmaceutical formulations and biological samples.
Testing of selectivity, repeatability, and reproducibility of 2.5Zn-Ca2CuO3/GCE
To ensure the selectivity of the fabricated electrode 2.5Zn-Ca2CuO3/GCE, influence of potential co-existing compounds such as uric acid, caffeine, theophylline, dopamine, sodium carbonate, starch, sodium chloride, potassium chloride, folic acid, and xanthine and some inorganic ions such as calcium, copper, iron, magnesium, and ammonium were examined by chronoamperometric method in the presence of AA, AP, and AM in 0.1 M PBS at a concentration ratio of 1:50 (Fig. S2A–C). It was noted that none of the investigated compounds have any effect on the amperometric response of AA, AP, and AM which demonstrates the excellent selectivity of the 2.5Zn-Ca2CuO3/GCE for the detection of AA, AP, and AM. The stability of the developed 2.5Zn-Ca2CuO3/GCE was investigated by sweeping the potential between 0.1 and 1.0 V at a step potential of 0.05 V/s in 0.1 M PBS (pH 7.0) containing 500 µM AA and 5 µM each AP and AM. The CVs of 50 cycles recorded at 2.5Zn-Ca2CuO3/GCE are shown in Fig. S2D. The voltammograms show that the anodic peak response for AA, AP, and AM oxidation changed to a small extent (5.3% for AA, 1.8% for AP, and 1.1% for AM) at each cycle, with a calculated recovery percentage of 85% for AA, 95% for AP, and 97% for AM.
To investigate the reproducibility of the developed 2.5Zn-Ca2CuO3/GCE for the sensitive determination of AA, AP, and AM, six different electrodes were fabricated under identical conditions and were used to determine AA, AP, and AM simultaneously. SWVs were measured in 0.1 M PBS (pH 7.0) containing 500 µM AA and 5 µM each AP and AM at six different 2.5Zn-Ca2CuO3/GCEs (Fig. S2E). The RSD value calculated for three replicative measurements was found to be 6.5% for AA, 2.6% for AP, and 1.3% for AM, demonstrating that the developed 2.5Zn-Ca2CuO3/GCE has superior reproducing ability. Based on these findings, it is proposed that the 2.5Zn-Ca2CuO3/GCE can be used for sensitive determination of AA, AP, and AM in pharmaceutical and biological samples where the electrode produces stable, reproducible results.
Real sample analysis
Determination of amlodipine in pharmaceutical tablet
In order to test the usefulness of the developed electrode, 2.5Zn-Ca2CuO3/GCE was applied for the determination of AM in pharmaceutical tablet. SWVs were recorded in 0.1 M PBS (pH 7.0) at 2.5Zn-Ca2CuO3/GCE (Fig. S2F). Upon addition of tablet solution, a strong and well-defined anodic peak at 0.77 V corresponding to AM was obtained. This peak gets increased with the increment of volume of tablet solution. It is observed that there were no other peaks except that of AM peak which confirmed that the other co-existing excipients (starch, sodium carbonate as denoted in the tablet strip) do not interfere with the AM determination. From the obtained results, the amount of AM present in the tablet was estimated to be 4.98 mg which is in good agreement with the labelled amount of 5 mg in the tablet. Moreover, the calculated values of recovery percentage and RSD are given in Table 2. Thus, the fabricated 2.5Zn-Ca2CuO3/GCE indicated its suitability for the determination of AM in pharmaceutical products.
Determination of AA, AP, and AM in human urine
In order to ensure the applicability of the sensor for clinical diagnosis, urine samples of the hypertension patients and healthy individuals have been investigated. Figure 6 depicts the SWVs measured in 0.1 M PBS (pH 7.0) containing hypertension patients’ urine at 2.5Zn-Ca2CuO3/GCE. When the urine sample was added, an oxidation peak for AA, AP, and AM were obtained at the potentials of ~0.2 V, 0.6 V, and 0.8 V, respectively, and the peak currents got increased with the addition of urine sample. In particular, samples 2–4 showed the peaks for AA (~0.2 V). Samples 2–7 exhibited the peaks for AP at ~0.6 V and the peak for AM appeared in the samples 1–2 and 4–7 (~0.8 V). The oxidation peak currents were observed to shift towards higher potentials upon the addition of larger volumes of urine samples. This phenomenon could be attributed to the matrix effect resulting from potential compounds present in the urine. From the obtained current values, the concentrations of AA, AP, and AM present in the urine samples was calculated (using Fig. 3) and the results are presented in Table ST2. It can be noticed that the calculated AA, AP, and AM ranged 9.5–16.7 µg/mL (AA), 0.7–4.74 µg/mL (AP), and 0.32–2.27 µg/mL (AM) in the investigated urine samples. This result clearly evidenced that certain percentages of unabsorbed AA, AP, and AM were excreted in the urine. Therefore, establishing the necessary dosage of medication for patients can contribute to cost reduction and prevent excessive utilization of drug molecules. In addition, urine sample of healthy individual (27 years old, male) was analyzed using the developed electrode (Fig. 6H) which did not show any response even for the addition of large volume urine sample into the cell containing 0.1 M PBS (pH 7.0). Hence, using the standard addition method, known amounts of AM were added to the cell containing urine. The developed electrode demonstrated good recovery percentages ranging from 99.5 to 100.2% in AM detection (Table ST3). Consequently, analyzing urine composition shall enable the clinicians to recommend an appropriate dosage for each individual. The sensor developed in this study proves beneficial for tailoring medicine to individual needs, supporting personalized healthcare. This remarkable result indicated the suitability of the electrode for the determination of AA, AP, and AM in clinical and pharmaceutical samples.

A–G SWVs obtained in seven hypertension patients’ human urine and (H) healthy individual urine at 2.5Zn-Ca2CuO3/GCE
Conclusion
The current study presents a simple, mediator-free nanoparticle-based electrode 2.5Zn-Ca2CuO3/GCE for the sensitive, selective, and simultaneous determination of amlodipine, acetaminophen, and ascorbic acid in pharmaceutical and urine samples. 2.5Zn-Ca2CuO3 nanoparticles were synthesized by chemical co-precipitation method and were characterized through XRD, XPS, VSM, and HRTEM. Subsequently, these nanoparticles dispersed in distilled water were drop casted onto the GCE surface, and the modified electrode was utilized to determine AA, AP, and AM with high precision. The 2.5Zn-Ca2CuO3 yielded promising results for the simultaneous determination of AA, AP, and AM with a linear concentration range of 50 to 3130 µM, 0.1 to 650 µM, and 0.15 to 378 µM and a LODs of 100 µM, 0.05 µM, and 0.07 µM, respectively. Additionally, the electrode demonstrated excellent stability, reproducibility, and repeatability, with acceptable RSDs and recovery percentages. Real-time application of the electrode was further explored by measuring the concentrations of AA, AP, and AM in pharmaceutical tablets and urine samples, both of which yielded high recovery percentages. Notably, the electrode proposed in this study had wider linear ranges and better LODs than the previous reported electrodes, making it a promising tool for pharmaceutical quality control and clinical applications.
Data availability
Data will be made available on request.
References
Jadon N, Jain R, Pandey A (2017) Electrochemical analysis of amlodipine in some pharmaceutical formulations and biological fluid using disposable pencil graphite electrode. J Electroanal Chem 788:7–13. https://doi.org/10.1016/j.jelechem.2017.01.055
Naikoo GA, Pandit UJ, Sheikh MUD et al (2020) Synergistic effect of carbon nanotubes, copper and silver nanoparticles as an efficient electrochemical sensor for the trace recognition of amlodipine besylate drug. SN Appl Sci 2:1–12. https://doi.org/10.1007/s42452-020-2807-z
Firouzi M, Giahi M, Najafi M et al (2020) Electrochemical determination of amlodipine using a CuO-NiO nanocomposite/ionic liquid modified carbon paste electrode as an electrochemical sensor. J Nanoparticle Res 23. https://doi.org/10.1007/s11051-021-05200-w
Atta NF, El-Ads EH, Galal A, Galal AE (2019) Electrochemical sensing platform based on nano-perovskite/glycine/carbon composite for amlodipine and ascorbic acid drugs. Electroanalysis 31:448–460. https://doi.org/10.1002/elan.201800577
Sudha K, Elangovan A, Jeevika A et al (2021) Electroanalytical detection of amlodipine in urine and pharmaceutical samples using Ag-Ce2(WO4)3@CNF nanocomposite-modified glassy carbon electrode. Microchem J 165:106138. https://doi.org/10.1016/j.microc.2021.106138
Švorc L, Cinková K, Sochr J et al (2014) Sensitive electrochemical determination of amlodipine in pharmaceutical tablets and human urine using a boron-doped diamond electrode. J Electroanal Chem 728:86–93. https://doi.org/10.1016/j.jelechem.2014.06.038
Madhuri C, Manohara Reddy YV, Prabhakar Vattikuti SV et al (2019) Trace-level determination of amlodipine besylate by immobilization of palladium-silver bi-metallic nanoparticles on reduced graphene oxide as an electrochemical sensor. J Electroanal Chem 847:113259. https://doi.org/10.1016/j.jelechem.2019.113259
Lou BS, Rajaji U, Chen SM, Chen TW (2020) A simple sonochemical assisted synthesis of NiMoO4/chitosan nanocomposite for electrochemical sensing of amlodipine in pharmaceutical and serum samples. Ultrason Sonochem 64:104827. https://doi.org/10.1016/j.ultsonch.2019.104827
Doulache M, Bakirhan NK, Saidat B, Ozkan SA (2020) Highly sensitive and selective electrochemical sensor based on polyglycine modified glassy carbon electrode for simultaneous determination of amlodipine and ramipril from biological samples. J Electrochem Soc 167:027511. https://doi.org/10.1149/1945-7111/ab68cd
Upreti V, Ratheesh VR, Dhull P, Handa A (2013) Shock due to amlodipine overdose. Indian J Crit Care Med 17:375–377. https://doi.org/10.4103/0972-5229.123452
Khairy M, Khorshed AA, Rashwan FA et al (2017) Sensitive determination of amlodipine besylate using bare/unmodified and DNA-modified screen-printed electrodes in tablets and biological fluids. Sens Actuators B Chem 239:768–775. https://doi.org/10.1016/j.snb.2016.07.165
Kang X, Wang J, Wu H et al (2010) A graphene-based electrochemical sensor for sensitive detection of paracetamol. Talanta 81:754–759. https://doi.org/10.1016/j.talanta.2010.01.009
Luo J, Fan C, Wang X et al (2013) A novel electrochemical sensor for paracetamol based on molecularly imprinted polymeric micelles. Sens Actuators B Chem 188:909–916. https://doi.org/10.1016/j.snb.2013.07.088
Solanki PR, Kaushik A, Agrawal VV, Malhotra BD (2011) Nanostructured metal oxide-based biosensors. NPG Asia Mater 3:17–24
Sun D, Zhang Y, Wang F et al (2009) Electrochemical sensor for simultaneous detection of ascorbic acid, uric acid and xanthine based on the surface enhancement effect of mesoporous silica. Sens Actuators B Chem 141:641–645. https://doi.org/10.1016/j.snb.2009.07.043
Zhao D, Yu G, Tian K, Xu C (2016) A highly sensitive and stable electrochemical sensor for simultaneous detection towards ascorbic acid, dopamine, and uric acid based on the hierarchical nanoporous PtTi alloy. Biosens Bioelectron 82:119–126. https://doi.org/10.1016/j.bios.2016.03.074
Wang C, Du J, Wang H et al (2014) A facile electrochemical sensor based on reduced graphene oxide and au nanoplates modified glassy carbon electrode for simultaneous detection of ascorbic acid, dopamine and uric acid. Sens Actuators B Chem 204:302–309. https://doi.org/10.1016/j.snb.2014.07.077
Atta NF, Galal A, Ahmed YM, El-Ads EH (2019) Design strategy and preparation of a conductive layered electrochemical sensor for simultaneous determination of ascorbic acid, dobutamine, acetaminophen and amlodipine. Sens Actuators B Chem 297. https://doi.org/10.1016/j.snb.2019.126648
Veerapandi G, Lavanya N, Sekar C (2023) Ca2CuO3 perovskite nanomaterial for electrochemical sensing of four different analytes in the xanthine derivatives family. Mater Chem Phys 295:127076. https://doi.org/10.1016/j.matchemphys.2022.127076
Lavanya N, Sekar C, Murugan R, Ravi G (2016) An ultrasensitive electrochemical sensor for simultaneous determination of xanthine, hypoxanthine and uric acid based on Co doped CeO2 nanoparticles. Mater Sci Eng C 65:278–286. https://doi.org/10.1016/j.msec.2016.04.033
Rosner H, Eschrig H, Hayn R et al (1997) Electronic structure and magnetic properties of the linear chain cuprates Sr2CuO3 and Ca2CuO3. Phys Rev B - Condens Matter Mater Phys 56:3402–3412. https://doi.org/10.1103/PhysRevB.56.3402
Zhu D, Ma H, Pang H et al (2018) Facile fabrication of a non-enzymatic nanocomposite of heteropolyacids and CeO2@Pt alloy nanoparticles doped reduced graphene oxide and its application towards the simultaneous determination of xanthine and uric acid. Electrochim Acta 266:54–65. https://doi.org/10.1016/j.electacta.2018.01.185
Pisoschi AM, Pop A, Serban AI, Fafaneata C (2014) Electrochemical methods for ascorbic acid determination. Electrochim Acta 121:443–460
El-Said WA, Nasr O, Soliman AIA et al (2021) Fabrication of polypyrrole/Au nanoflowers modified gold electrode for highly sensitive sensing of paracetamol in pharmaceutical formulation. Appl Surf Sci Adv. https://doi.org/10.1016/j.apsadv.2021.100065. 4:
Dehdashti A, Babaei A (2020) Highly sensitive electrochemical sensor based on Pt doped NiO nanoparticles/MWCNTs nanocomposite modified electrode for simultaneous sensing of piroxicam and amlodipine. Electroanalysis 32:1017–1024. https://doi.org/10.1002/elan.201900580
Atta NF, Galal A, El-Gohary ARM (2022) Electrochemical sensing of dobutamine, paracetamol, amlodipine, and daclatasvir in serum based on thiourea SAMs over nano-gold particles-CNTs composite. New J Chem 46:12265–12277. https://doi.org/10.1039/d2nj01822e
Eskiköy Bayraktepe D, Yazan Z (2020) Application of single-use electrode based on nano-clay and MWCNT for simultaneous determination of acetaminophen, ascorbic acid and acetylsalicylic acid in pharmaceutical dosage. Electroanalysis 32:1263–1272. https://doi.org/10.1002/elan.201900601
Akbari Javar H, Mahmoudi-Moghaddam H, Garkani-Nejad Z, Dehghannoudeh G (2022) Grass-like Pt-doped NiCo2O4 modified electrode for electrochemical detection of amlodipine. Meas J Int Meas Confed 191. https://doi.org/10.1016/j.measurement.2022.110790
Deng P, Feng J, Xiao J et al (2021) Application of a simple and sensitive electrochemical sensor in simultaneous determination of paracetamol and ascorbic acid. J Electrochem Soc 168:096501. https://doi.org/10.1149/1945-7111/ac1e59
Funding
The authors acknowledge DST sponsored Indo-Sri Lanka project (DST/INT/SL/P-30/2021) and MHRD-RUSA 2.0 (No. F.24–51/2014-U, Policy (TN-Multi-Gen), dated: 09.10.2018) for financial assistance. The authors also thank Mr. S. Arunachalam for many stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
Urine samples for this research have been collected from the Government Hospital in Karaikudi, where doctors conduct clinical tests that meet ethical norms and protect participants’ rights and well-being. As a result, ethical approval may not always be necessary for the use of human urine samples.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Veerapandi, G., Sekar, C. Binder-free and efficient voltammetric sensor based on Zn-Ca2CuO3 nanoparticles for simultaneous determination of amlodipine, acetaminophen, and ascorbic acid in hypertension patients. Microchim Acta 191, 409 (2024). https://doi.org/10.1007/s00604-024-06473-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-024-06473-3