
Abstract
The authors describe an electrochemiluminescent (ECL) DNA biosensor that is based on the use of gold nanoparticles (AuNPs) modified with graphite-like carbon nitride nanosheets (g-C3N4 NSs) and carrying a DNA probe. In parallel, nanoparticles prepared from gold-platinum (Au/Pt) alloy and carbon nanotubes (CNTs) were placed on a glassy carbon electrode (GCE). Once the g-C3N4 NHs hybridize with DNA-modified AuNPs, they exhibit strong and stable cathodic ECL activity. The Au/Pt-CNTs were prepared by electrochemical deposition of Au/Pt on the surface of the CNTs in order to warrant good electrical conductivity. On hybridization of immobilized capture probe (S1), target DNA (S2) and labeled signal probe (S3), a sandwich-type DNA complex is formed that produces a stable ECL emission at a typical applied voltage of −1.18 V and in the presence of peroxodisulfate. Under optimized conditions, the method has a response to target DNA that is linearly related to the logarithm of its concentration in the range between 0.04 f. and 50 pM, with a 0.018 f. detection limit.

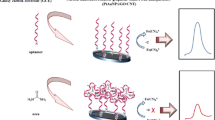
Schematic presentation of an electrochemiluminescent DNA biosensor based on two-dimensional graphite-like carbon nitride nanosheets (g-C3N4 NSs) hybridized with gold nanoparticles. Abbreviations: MCH: 6-mercapto-1-hexanol; S1: Capture probe; S2: target DNA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pursuit of rapid and sensitive detection of specific DNA assay has received significant attention, due to its ever increasing demands for genetic diseases, disorders, the detection and characterization of viruses, bacteria, and parasites [1–3]. To realize the specific and sensitive DNA detection, numerous systems have been established including chemiluminescence (CL), fluorescence, atomic force microscopy, surface plasmon resonance spectroscopy, electrochemical techniques, and so forth [4–9]. Among the diverse techniques, electrochemiluminescence (ECL) integrating the merits of chemiluminescence and electrochemistry, has achieved overwhelming success because of its extremely high sensitivity, wide dynamic range, rapidness, simplicity, and stable labels by coupling with immunoassays and DNA probe assays.
2D nanomaterials, consisting of single or few atomic layers, have attracted tremendous attention due to their extraordinary properties and potential applications [10–14]. Graphite-like carbon nitride (g-C3N4) as a typical applicable 2D material semiconductor consists of carbon and nitrogen, which exhibits structural similarity to graphene but different from the property point of view, has received great attention [15]. Investigations on single or several-atom-thick g-C3N4 nanosheets (NSs) have proven their potential with respect to high water-dispersibility, high fluorescent quantum yield, strong electrochemiluminescence (ECL) activity, good stability, and improved photocatalytic property compared to their bulk counterparts [16–18].
However, it was reported that the ECL stability of g-C3N4 NSs significantly decreases with the increasing of applied potential; as the scanning potential beyond −0.9 V, over-injection of highly energetic electrons into the conduction bands of g-C3N4 NSs resulted in the partial electro-reduction of g-C3N4 NSs to form a less conductive layer, eventually blocks subsequent ECL emissions, which is distinctly unfavorable for the construction of reusable and sensitive ECL biosensor based on g-C3N4 NSs [19]. The passivation problem of g-C3N4 NSs can be resolved by functionalizing supported metal nanoparticles (MNPs) with optimized size and shape, which results in controlled access to metal-semiconductor heterojunctions and enhanced electron transfer between photoexcited semiconductor and MNPs [20].
Herein, AuNPs were deposited on both sides of g-C3N4 NSs and form a sandwich-like Au-g-C3N4 nanocomposite. As a consequence, the prepared Au-g-C3N4 NSs exhibit strong and stable cathodic ECL activity compared to g-C3N4 NSs due to the important roles of AuNPs in trapping and storing the electrons from the conduction band of g-C3N4 NSs, as well as preventing high energy electron induced passivation of g-C3N4 NSs. In addition, AuNPs can prevent g-C3N4 NSs from being electrochemically degraded and catalyze the reduction of persulfate (S2O8 2−) into hole donor (SO4 ·-) at the same time, thus leading to the enhancement of ECL emission of Au-g-C3N4 NSs nanohybrids.
Carbon nanotubes (CNTs), pseudo-one-dimensional carbon allotropes, possess high aspect ratio, high surface area, and excellent material properties [21], that make them unique properties for applications in various fields such as electronics, catalysis, and medicine [22]. To expand CNTs potential applications, strategies for functionalizing CNTs by doping or grafting are critical for the pursuit of making a desirable platform for various applications. Noble metal nanoparticles possess the merits of high surface reactivity, good solubility, and excellent bioactivity [23]. Furthermore, bimetallic nanoalloys nanoparticles have also been demonstrated to possess both synergistic effect and electronic effect which are distinctly superior to those of their monometallic counterparts. And noble metal nanoparticles attached to or deposited on the CNTs have drawn considerable attention owing to their widespread applications [24]. Herein, a technique has emerged to integrate CNTs with Au/Pt alloy NPs and further applied in the fabrication of DNA sensing platform, which can not only improve the surface area, but also facilitate the electron transfer for signal amplification.
Herein, we report a novel ECL strategy for high sensitive detection of specific sequences DNA based on Au/Pt-CNTs as substrate combined with the excellent ECL activity of Au-g-C3N4 NSs. In the presence of target DNA (S2), the Au-g-C3N4 NSs labeled signal probe (S3) can hybridize with S2 and produce a remarkable amplified ECL signal in the presence of S2O8 2−. With the increasing of S2, more Au-g-C3N4 NSs labeled S3 was immobilized on the electrode, leading to ascend of the ECL intensity. On the basis of variation of the ECL intensity, a novel ECL DNA biosensor was constructed. This designed DNA biosensor allowed the rapid detection of this target with a dynamic range and excellent selectivity, which makes it a potential for various applications in highly sensitive determination of DNA.
Experimental section
Reagents
Carbon nanotubes (CNTs, CVD method, purity≧ 98%) were purchased from Nanoport Co. Ltd. (http://www.nanotubes.com.cn/) (Shenzhen, China). The ultrapure water was obtained from a Lichun water purification system (≥18 MΩ·cm−1) and used throughout. 6-mercapto-1-hexanol (MCH) was purchased from Nanoport. Co. Ltd. (http://www.nanotubes.com.cn/) (Shenzhen, China). The buffers involved in this work are as follows: DNA immobilization buffer, 10 mM Tris-HCl and 0.1 M NaCl (pH 7.4); hybridization buffer, 10 mM phosphate buffered saline (PBS, pH 7.4) with 0.25 mM NaCl; washing buffer, 10 mM PBS, and 0.1 mM NaCl (pH 7.4). 10 mM Tris-HCl buffer (pH 7.4) containing 0.1 M K2S2O8 and 0.1 M KCl were used as the coreactant for ECL system. Guanidine hydrochloride and hydrogen hexachloroplatinate (IV) hydrate (H2PtCl6·6H2O) were obtained from Sigma-Aldrich (www.sigmaaldrich.com), chloroauric acid (HAuCl4·4H2O) was obtained from Shanghai Reagent Company (http://www.alibaba.com/showroom/shanghai-chemical-reagent-co.html) (Shanghai, China), and all the other reagents were at least of analytical grade.
The human serum samples were provided by Shandong Tumor Hospital and those DNA sequences are presented below.
-
Capture DNA (S1): 5′-TGG AAA ATC TCT AGC AGT CGT-(CH2)6-NH2–3′
-
Target DNA (S2): 5′-ACT GCT AGA GAT TTT CCA CAC TGACTA AAA GGG TCT GAG GGA-3′
-
Signal DNA (S3): 5′-SH-(CH2)6-ATG TCC CTC AGA CCCTTT-3′
-
One-base mismatched DNA (S4): 5′-ACT GCT AGA GAT TTT CCA CAC TGA CTA AAA GGG TCT GTG GGA-3
-
Non-complementary (S5): 5′-ACT GCT AGA GAT TTTCCA CAC TGA CTA CTT CAA CAG TGC CCC-3′
Apparatus
The ECL measurements were conducted on a flow injection luminescence analyzer (IFFM-E, Xi’an Remex Electronic Instrument High-Tech Ltd., Xi’an, China) with the voltage of the photomultiplier tube (PMT) set at 800 V. Cyclic voltammetric measurements (CVs) were performed with a CHI760D electrochemical workstation (Shanghai CH Instruments, China). Transmission electron microscopy (TEM) images were obtained from a Hitachi H-800 microscope (Japan). Electrochemical impedance spectroscopy (EIS) was carried out on an IM6x electrochemical station (Zahner, Germany). Scanning electron microscope (SEM) images were obtained using a QUANTA FEG 250 thermal field emission SEM (FEI Co., USA). Energy dispersive spectrometer (EDX) was obtained using an Oxford X-MAX50 EDX (Oxford, Britain). Ultraviolet visible (UV-vis) was recorded on a UV-3101 spectrophotometer (Shimadzu, Japan). The photoluminescence characterization was achieved on a LS-55 spectrofluorometer (P. E. USA).
Preparation of acidized CNTs
The commercially purchased CNTs were purified and oxidized according to the literature with some modifications [25]. First, the CNTs were pretreated by refluxing CNTs in 60% HNO3 at 60 °C for 6 h to remove the metal particles and other impurities. After the purification process, the surface oxidation of the CNTs was carried out by 100 mL of a HNO3/H2SO4 (1/3) mixture in an ultrasonic bath for 12 h at room temperature. Next, the acid-treated CNTs was separated and washed by centrifugation with doubly distilled water to neutral pH. After treatment with a 0.22 μm fitration membrane to remove overlong CNTs, the oxidized CNTs were dried in an oven at 60 °C overnight, and finally stored in water at 2 mg·mL−1 for further use. The presence of carboxylic groups on the surface of acidoxidized CNTs provides a negative surface charge. The acidic treatment has the secondary effect of making the CNTs shorter and less entangled, leading to suspensions of well-dispersed individual, charged nanotube segment.
Synthesis of g-C3N4 NSs
The bulk g-C3N4 NSs were prepared following the previously reported literature [26]. Typically, analytical grade guanidine hydrochloride (4 g) was put into an alumina crucible with a cover and placed in the middle region of the quartz tube (25 mm in inner diameter and 1000 mm in length). In order to react adequately, the initial materials were heated in an electric furnace at a rate of 2.5 °C·min−1 up to 450 °C, and this temperature was maintained for 3 h under ambient atmospheric conditions. The quartz tube was then cooled to ambient temperature. After that, the yellow g-C3N4 NSs powders were collected in the covered crucible. Then, the g-C3N4 NSs were obtained by liquid exfoliation of prepared bulk g-C3N4 in water. Briefly, 100 mg of bulk g-C3N4 NSs powder was placed into a mortar, then dispersed into 100 mL water and ultrasonic treated for 30 min. The unexfoliated g-C3N4 was separated through centrifugation at about 6000 rmp, the supernatant was concentrated with a rotary evaporator at 60 °C under reduced pressure. And a milk-like suspension with the concentration of about 2 mg·mL−1 was obtained.
Preparation of the Au-g-C3N4 NSs
To prepare the Au-g-C3N4 NSs, 10 μL of 0.01 M HAuCl4 solution and 2 mL of the above prepared g-C3N4 NSs suspension were mixed under stirring. After 10 min sonication with 2 min interval for 3 times, 25 μL of 0.01 M freshly prepared NaBH4 solution was added quickly to the suspension to reduce the AuCl4 −, followed by continuously stirring for another 20 min. After that, 10 μL of 0.01 M sodium citrate solution was dropped into above solution and continued for 30 min stirring. The Au-g-C3N4 NSs possessed good dispersibility and stability was obtained with a concentration about 2 mg·mL−1 after remove excess NaBH4, sodium citrate and unbound gold nanoparticles by centrifugation.
Preparation of Au-g-C3N4 NSs conjugated signal DNA (S3)
The prepared Au-g-C3N4 NSs composites were first mixed with S3 solution (1 mL, 50 μM). After incubating for 2 h at 4 °C with gentle stirring, the mixture was washed with buffer, and centrifuged (7000 rpm, 3 min) for three times. Following that, the precipitation was diluted into 0.5 mL with PBS further blocked possible remaining active sites with 1.0% (wt) MCH. At last, the Au-g-C3N4 NSs labeled S3 conjugation (S3-Au-g-C3N4 NSs) were aged in salts solution (0.1 M NaCl and 10.0 mM acetate buffer) for 24 h, followed by centrifuging at 10,000 rpm for 10 min. The composites were added into immobilization buffer and stored at 4 °C prior to use.
Fabrication of the DNA biosensor
The DNA biosensor was prepared as the following steps which can be seen from Scheme 1: (i) prior to surface modification, the glassy carbon electrode (GCE) (5 mm diameter) was firstly polished with alumina slurry and rinsed thoroughly with absolute alcohol and distilled water in ultrasonic bath, and dried in air at room temperature; (ii) 10 μL of CNTs composites were initially deposited on the electrode surface, and then dried at room temperature; (iii) the electrochemical deposition of Au/Pt was performed on the CNTs modified GCE in aqueous solution containing 1.0 mM HAuCl4 and 1.0 mM H2PtCl6, with the deposition time 200 s and the potential −0.2 V; (iv) after washed with PBS (pH 7.4), the GCE was immersed into an immobilization buffer containing 10 nM capture probe DNA (S1) for 40 min, S1 were effectively assembled on the surface of Au/Pt-CNTs composites modified electrode; (v) the S1 modified electrode was further treated with 1.0% (wt) MCH for 30 min to obtain a well aligned DNA monolayer and block the possible remaining active sites, followed by washing with water to remove physical adsorption; (vi) for the hybridization reaction, the modified electrode was immersed into stirred Tris-HCl solution containing varying concentration of S2 for 2 h at 42 °C allowing DNA strands to be stretched out well, which was an important procedure (before hybridization with the capture probe was denatured to single-stranded DNA by keeping the tube at 95 °C for 10 min and then in a freezing mixture consisting NaCl and ice for 1 min); (vii) the electrode was hybridized with signal reporter (S3-Au-g-C3N4 NSs bioconjugates) for 2 h at 37 °C. After hybridization, the electrode was extensively rinsed with washing buffer (10 mM Tris-HCl, pH 7.4) and dried under a stream of nitrogen prior to characterization. The ECL emission was detected by the PMT biased at 800 V. The ECL signals related to the S2 concentrations were measured as well.

Construction process of the ECL DNA sensing strategy
Results and discussion
Characterization of CNTs and au/Pt-CNTs
The surface morphology of the CNTs and Au/Pt-CNTs composites was detailedly verified by SEM and energy dispersive spectrometer (EDS) analysis, shown in Fig. 1. The carboxylated CNTs obtained by chemical oxidation shows a homogeneous surface and good dispersion (Fig. 1a). Compared with Fig. 1a, it is observed in Fig. 1b that the nanoparticles decorated uniformly on the surface of CNTs. Furthermore, the EDS analysis is also used to evaluate the surface functionalization by Au/Pt (Fig. 1a).

SEM image of CNT a and Au/Pt-CNTs composites b; c EDS analysis of Au/Pt-CNTs composites
Characterization of g-C3N4 NSs and Au-g-C3N4 NSs
The prepared g-C3N4 NSs and Au-g-C3N4 NSs were characterized by TEM, XRD, UV-vis absorption spectra and photoluminescence (PL) spectroscopy. It is clearly observed that g-C3N4 NSs was irregular thick block material with a two-dimensional sheet-like structure (Fig. 2a). Compared with g-C3N4 NSs, large numbers of black colored dots corresponding to the AuNPs uniformly distributed on the surface of g-C3N4 NSs without agglomeration (Fig. 2b). The crystallinity and phase composition of the synthesized g-C3N4 NSs and Au-g-C3N4 NSs was investigated by XRD (Fig. 2c). There is a characteristic diffraction peak at 27.2° (curve a in Fig. 2c) associated with the aromatic systems interplanar stacking of g-C3N4 NSs [16]. After the formation of Au-g-C3N4 NSs, two new diffraction peaks of 38.2° and 44.4°, typical peaks for the (111) and (200) planes of AuNPs, are observed (curve b in Fig. 2c), indicating the deposition of AuNPs on the g-C3N4 NSs. In the UV-vis absorption spectra (Fig. 2d), compared with g-C3N4 NSs alone (curve a), an obscure absorption peak around 520 nm, the characteristic absorption peak of spherical AuNPs, is observed, further demonstrated the formation of Au-g-C3N4 NSs (curve b). Both g-C3N4 NSs (curve c) and Au-g-C3N4 NSs (curve d) displayed a luminescence peak at about 450 nm, Inset in Fig. 2d shows that the g-C3N4 NSs (middle) and Au-g-C3N4 NSs (right) display blue fluorescence exposed to UV light of 365 nm wavelength.

TEM image of g-C3N4 NSs a and Au-g-C3N4 NSs b; XRD patterns c of g-C3N4 NSs (curve a) and Au-g-C3N4 NSs (curve b); d UV-vis absorption spectra (black line) and PL spectroscopy (red line) of g-C3N4 NSs (curve a and c) and Au-g-C3N4 NSs (curve b and d), inset shows in Fig. 2d the color change of g-C3N4 NSs solution without (left) and with UV irradiation (middle) and Au-g-C3N4 NSs (right) exposure to UV irradiation
EIS characterization of the DNA biosensor
EIS is an effective method to monitor the changes of interfacial properties, allowing the understanding of chemical transformation and processes associated with the conductive electrode surface. Electrochemical impedances of the electrodes were performed in a background solution of 5.0 mM K3Fe(CN)6 containing 0.1 M KCl, and the frequency range is at 100 mHz to 10 kHz at a bias potential of 170 mV (vs Ag/AgCl). In EIS, the diameter of the semicircle at higher frequencies corresponds to the electron-transfer resistance (R et ); a change in the value of R et was associated with the blocking behavior of the modification processes in the GCE, and was reflected in the EIS as a change in the diameter of the semicircle at high frequencies. Figure 3a shows the EIS corresponding to the stepwise modification of the GCE at different stages. The R et of bare GCE was small (curve a). When CNTs was modified on the GCE surface, the R et decreased markedly owing to the superior electroconductibility of CNTs (curve b). It was observed that the R et further decreased after deposition of Au/Pt on the surface of CNTs, indicating that the Au/Pt-CNTs is an excellent electric conducting material and accelerated the electron transfer (curve c). Significant gradual increases in R et values after immobilized S1 and blocked with MCH onto the electrode surface were observed, which was attributed to the insulating property of protein molecules (curve d). The R et increased continuously (curves e and f) after the DNA biosensor incubated with target DNA S2 and S3-Au-g-C3N4 NSs, suggesting successful fabrication of the biosensor. Electrical equivalent circuit (inset in Fig. 3a) was initiated to fit satisfactorily the data as given in Table 1 obtained over the measured frequency range.

a EIS response of (a) bare GCE, b CNT modified GCE, c electro-deposition of Au/Pt on CNT modified GCE, d after S1 and MCH modification, e hybided with target DNA S2, f immobilization of S3-Au-g-C3N4 NSs, Inset: the electrical equivalent circuit applied to fit the impedance data; Rs the ohmic resistance of the electrolyte solution, Ret is the electron transfer resistance, Rf film resistance, Q1 is the constant phase element related to the charge capacitance at the electrode/electrolyte interface, Q2 is the constant phase element related to the charge capacitance at the metal/electrolyte interface and W is the Warburg impedance due to mass transfer to the electrode surface; (B) ECL responses of g-C3N4 NSs (2 mg·mL−1) and Au-g-C3N4 NSs (2 mg·mL−1) modified Au/Pt-CNTs decorated GCE in the absence (curve a and b) and presence (curve c and d) of S2O8 2−
ECL emission of the DNA biosensor
The possible ECL reaction mechanisms are presented in Electronic Supporting Material (ESM). Figure 3b displays the ECL curves of the g-C3N4 NSs and Au-g-C3N4 NSs-modified GCE in the absence (curve a and c) and presence (curve b and d) of coreactant S2O8 2− by cycling the potential between 0 and 1.5 V. It can be seen that there is a weak cathodic ECL signal response on the g-C3N4 NSs and Au-g-C3N4 NSs-modified GCE in the buffer without S2O8 2− solution due to the weak ECL reaction between g-C3N4 NSs and Au-g-C3N4 NSs and dissolved oxygen (curve a and b). However, both g-C3N4 NSs and Au-g-C3N4 NSs exhibited bright ECL emission in the presence of S2O8 2− (curve c and d), which ascribed to the strong high-energy annihilation between electrons and holes [27]. The Au-g-C3N4 NSs has a higher ECL emission (curve d) compared with g-C3N4 NSs (curve c), lower ECL onset potential (−0.90 V), and peak potential (−1.18 V) that arise from AuNPs electrocatalytic reduction toward S2O8 2− and more hole-donor (SO4·− free radical) generated [19].
To demonstrated that AuNPs play an important role in stabilizing ECL intensity of g-C3N4 NSs, the ECL emission of g-C3N4 NS film and Au-g-C3N4 NSs were investigated. As shown in Fig. 4a, in the presence of coreactant K2S2O8 with an applied potential scanning from 0 to −1.5 V, the ECL emission of bare g-C3N4 NS rapidly degrades with cyclic potential scan increased, which was ascribed to electrode passivation occurring after cyclic potential scan, leading to a sharp decrease in ECL emission at subsequent potential scans [19]. While, the ECL emission from Au-g-C3N4 NSs is quite stable under 10 repeated cyclic voltammetric scans (Fig. 4b), indicating its satisfying reversibility and reliability as a sensing signal.

ECL emission from g-C3N4 NS film a and Au-g-C3N4 NSs b under continuous CVs for 10 cycles with applied potential from −1.5 to 0 V. The scan rates were 100 mV·s−1
To monitor the effect of Au/Pt-CNTs on the signal amplification of the ECL bioassay, four types of substrate (CNTs, Au-CNTs, Pt-CNTs and Au/Pt-CNTs) were prepared. As shown in Fig. 5, the Au-g-C3N4 NSs modified Au/Pt-CNTs decorated GCE (curve e) showed maximum ECL emission compared with other substrate (curve b-d) ascribed to the formation of excellent conductive layers on the surface of the GCE. No ECL emission was observed in the absence of Au-g-C3N4 NSs (curve a). The results indicated that the origination of the ECL response was from Au-g-C3N4 NSs with K2S2O8 as coreactant.

ECL responses of Au/Pt-CNTs decorated GCE (curve a), Au-g-C3N4 NSs modified CNTs decorated GCE (curve b), Au-g-C3N4 NSs modified Pt-CNTs decorated GCE (curve c); Au-g-C3N4 NSs modified Au-CNTs decorated GCE (curve d), and Au-g-C3N4 NSs modified Au/Pt-CNTs decorated GCE containing 0.1 M K2S2O8. The PBS was 0.1 M phosphate with pH 7.4. The scan rates were all 100 mV·s−1. The voltage of the PMT was set as 800 V
Analytical performance
Prior to DNA quantification, the following parameters were optimized: (a) Sample pH value; (b) Incubation time; (c) Incubation temperature. Respective data and Figures are given in the Electronic Supporting Material. We found the following experimental conditions to give best results: (a) A sample pH value of 7.4; (b) Incubation time 2 h; (c) Incubation temperature 37 °C.
The quantitative behavior of the DNA biosensor was assessed by monitoring the difference of the ECL intensity upon the concentration of S2 at optical conditions. As shown in Fig. 6, the ECL intensity of the sensor increased with the increasement of S2 concentrations, and exhibited a good linear relationship with the logarithm of S2 concentration from 0.04 f. to 50 pM. The linear regression equation was I ECL = 887.71 + 360.52 lg(cDNA/fM), with a correlation coefficient of 0.9966. The detection limit value for S2 was determined at 0.018 fM, estimated at a signal-to-noise of 3 criterion, which suggested a satisfactory detection limit and linear range. Compared with some existing DNA detection methods, this ECL biosensor using Au-g-C3N4 NSs as probe, provides a more effective approach with wider dynamic concentration response range and lower detection limit for the analysis of DNA (Table 2).

Calibration curve corresponding to the ECL signals as a function of target DNA S2 concentration. Inset shows the linear relationship between the ECL signals and the logarithm of the target DNA S2 concentration
Specificity, stability and reproducibility of the DNA sensor
The specificity of the designed DNA sensor in discriminating perfect target DNA from one-base mismatched (S4) and non-complementary (S5) was tested via comparing the ECL signal changes. The results demonstrated that this DNA biosensor exhibited an excellent specificity to the target DNA (Fig. 7a). This result suggests that the ECL biosensor has good ability of distinguishing mismatched DNA, and can be implemented for single nucleotide polymorphism detection. Stability of the DNA sensor is a key factor in their application. As shown in Fig. 7b, the ECL signal had little change under continuous potential scanning for 10 cycles, which indicated the good storage stability of the DNA biosensor. To investigate the reproducibility of the DNA sensor, intra- and inter-assay were both estimated. The reproducibility expressed as relative standard deviation was 4.7% for intra-assay and 3.8% for inter-assay. The experimental results indicated that this strategy was reliable and can be used for target DNA detection with acceptable reproducibility.

a Specificity of this ECL DNA biosensor in detecting different targets; b ECL response of the DNA biosensor with 10 f. target DNA under a continuous cyclic potential scan between 0 and −1.5 V for 10 cycles at 100 mV·s−1
Practical application
To demonstrate the analytical reliability of this method for clinical application, eleven replicate determinations of target DNA in spiked human serum samples were carried out under the optimal condition. Different amount of target DNA (10 fM, 100 fM, 500 fM, 1000 fM) were added into the spiked human serum sample. Results were displayed in Table 3 with a satisfactory recovery in the range from 97.3–104.7%. The results demonstrate the ability of our ECL DNA biosensor to detect perfect matches in human serum, which would be expended for accurate gene diagnostics.
Conclusions
In summary, we design a new ECL detection platform for ultrasensitive monitoring of DNA based on Au-g-C3N4 NSs as the signal-transduction tags. Introduction of gold nanoparticles greatly strengthened the ECL emission with the existence of coreactant K2S2O8 ascribed to the efficience catalyzing to S2O8 2− and reducing electron over-injection, and enhanced the detectable sensitivity of this biosensor. In addition, the combination of Au/Pt-CNTs produced a synergic effect in the analytical performance of the resulting electrode, which can improve the electronic transmission rate as well as increase the surface area. The proposed biosensor performed good sensitivity and accuracy for the detection of target DNA. This study offers an alternative, simple, and cost-effective technique for trace target DNA molecular, also holds great potential for point-of-care testing, public health and environmental monitoring in remote regions, developing or developed countries.
References
Lam B, Das J, Holmes RD, Live L, Sage A, Sargent EH, Kelley SO (2013) Solution-based circuits enable rapid and multiplexed pathogen detection Nat Commun 4. doi:10.1038/ncomms3001
Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW, Alizadeh AA, Diehn M (2014) An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20:552–558. doi:10.1038/nm.3519
Hsieh K, Patterson AS, Ferguson BS, Plaxco KW, Soh HT (2012) Rapid, sensitive, and quantitative detection of pathogenic DNA at the point of care through microfluidic electrochemical quantitative loop-mediated isothermal amplification. Angew Chem 124:4980–4984. doi:10.1002/ange.201109115
Freeman R, Liu X, Willner I (2011) Chemiluminescent and chemiluminescence resonance energy transfer (CRET) detection of DNA, metal ions, and aptamer–substrate complexes using hemin/G-quadruplexes and CdSe/ZnS quantum dots. J Am Chem Soc 133:11597–11604. doi:10.1021/ja202639m
He S, Song B, Li D, Zhu C, Qi W, Wen Y (2010) A graphene nanoprobe for rapid, sensitive, and multicolor fluorescent DNA analysis. Adv Fun Mater 20:453–459. doi:10.1002/adfm.200901639
Han WH, Liao JM, Chen KL, Wu SM, Chiang YW, Lo ST (2010) Enhanced recognition of single-base mismatch using locked nucleic acid-integrated hairpin DNA probes revealed by atomic force microscopy nanolithography. Anal Chem 82:2395–2400. doi:10.1021/ac902665c
Homola J (2008) Surface plasmon resonance sensors for detection of chemical and biological species. Chem Rev 108:462–493. doi:10.1021/cr068107d
Hu L, Xu G (2010) Applications and trends in electrochemiluminescence. Chem Soc Rev 39:3275–3304. doi:10.1039/B923679C
Cosnier S, Mailley P (2008) Recent advances in DNA sensors. Analyst 133:984–991. doi:10.1039/B803083A
Huang X, Zeng Z, Fan Z, Liu J, Zhang H (2012) Graphene-based electrodes. Adv Mater 24:5979–6004. doi:10.1002/adma.201201587
Xiang Q, Yu J, Jaroniec M (2012) Graphene-based semiconductor photocatalysts. Chem Soc Rev 41:782–796. doi:10.1039/C1CS15172J
Zhu Y, Murali S, Stoller MD, Ganesh KJ, Cai W, Ferreira PJ, Pirkle A, Wallace RM, Cychosz KA, Thommes M, Su D, Stach EA, Ruoff RS (2011) Carbon-based supercapacitors produced by activation of grapheme. Science 332:1537–1541. doi:10.1126/science.1200770
Xu C, Xu B, Gu Y, Xiong Z, Sun J, Zhao XS (2013) Graphene-based nanocomposites: preparation, functionalization, and energy and environmental applications. Energy Environ Sci 6:1388–1414. doi:10.1039/C3EE42518E
Xu HH, Wang YZ, Hu SH (2017) Nanocomposites of graphene and graphene oxides: synthesis, molecular functionalization and application in electrochemical sensors and biosensors. A Review doi:10.1007/s00604-016-2007-0
Wang Y, Wang XC, Antonietti M (2012) Polymeric graphitic carbon nitride as a heterogeneous organocatalyst: from photochemistry to multipurpose catalysis to sustainable chemistry. Angew Chem Int Ed 51:68–89. doi:10.1002/anie.201101182
Zhao XD, Xie X, Wang H, Zhang JJ, Pan BC, Xie Y (2013) Enhanced photoresponsive ultrathin graphitic-phase C3N4 nanosheets for bioimaging. J Am Chem Soc 135:18–21. doi:10.1021/ja308249k
Yang SB, Gong YJ, Zhang JS, Zhan L, Ma LL, Fang ZY, Vajtai R, Wang XC, Ajayan PM (2013) Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light. Adv Mater 25:2452–2456. doi:10.1002/adma.201204453
Chen LC, Huang DJ, Ren SY, Dong TQ, Chi YW, Chen GN (2013) Preparation of graphite-like carbon nitride nanoflake film with strong fluorescent and electrochemiluminescent activity. Nano 5:225–230. doi:10.1039/C2NR32248J
Chen LC, Zeng XT, Si P, Chen YM, Chi YW, Kim DH, Chen GN (2014) Gold nanoparticle-graphite-like C3N4 nanosheet nanohybrids used for electrochemiluminescent immunosensor. Anal Chem 86:4188–4195. doi:10.1021/ac403635f
Li XH, Wang XC, Antonietti M (2012) Mesoporous g-C3N4 nanorods as multifunctional supports of ultrafine metal nanoparticles: hydrogen generation from water and reduction of nitrophenol with tandem catalysis in one step. Chem Sci 3:2170–2174. doi:10.1039/C2SC20289A
Zhen SJ, Chen LQ, Xiao SJ, Li YF, Hu PP, Zhan L, Peng L, Song EQ, Huang CZ (2010) Carbon nanotubes as a low background signal platform for a molecular aptamer beacon on the basis of long-range resonance energy transfer. Anal Chem 82:8432–8437. doi:10.1021/ac100709s
Zhang Q, Huang JQ, Qian WZ, Zhang YY, Wei F (2013) The road for Nanomaterials industry: a review of carbon nanotube production, post-treatment, and bulk applications for composites and energy storage. Small 9:1237–1265. doi:10.1002/smll.201203252
Xu W, Xue XJ, Li TH, Zeng HQ, Liu XG (2009) Ultrasensitive and selective colorimetric DNA detection by nicking endonuclease assisted nanoparticle amplification. Angew Chem Int Ed 48:6849–6852. doi:10.1002/anie.200901772
Lollmahomed FB, Narain R (2011) Photochemical approach toward deposition of gold nanoparticles on functionalized carbon nanotubes. Langmuir 27:12642–12649. doi:10.1021/la2025268
Ajayan PM (1999) Nanotubes from carbon. Chem Rev 99:1787. doi:10.1021/cr970102g
Tang YR, Song HJ, Su YY, Lv Y (2013) Turn-on persistent luminescence probe based on graphitic carbon nitride for imaging detection of Biothiols in biological fluids. Anal Chem 85:11876–11884. doi:10.1021/ac403517u
Miao WJ (2008) Electrogenerated chemiluminescence and its biorelated applications. Chem Rev 108:2506–2553. doi:10.1021/cr068083a
Zhang SS, Zhong H, Ding CF (2008) Ultrasensitive flow injection Chemiluminescence detection of DNA hybridization using signal DNA probe modified with au and CuS nanoparticles. Anal Chem 80:7206–7212. doi:10.1021/ac800847r
Gerion D, Chen FQ, Kannan B, Fu AH, Parak WJ, Chen DJ, Majumdar A, Alivisatos AP (2003) Room-temperature single-nucleotide polymorphism and multiallele DNA detection using fluorescent nanocrystals and microarrays. Anal Chem 75:4766–4772. doi:10.1021/ac034482j
Zhang DD, Peng YG, Qi HL, Gao Q, Zhang CX (2010) Label-free electrochemical DNA biosensor array for simultaneous detection of the HIV-1 and HIV-2 oligonucleotides incorporating different hairpin-DNA probes and redox indicator. Biosens Bioelectron 25:1088–1094. doi:10.1016/j.bios.2009.09.032
Sato K, Hosokawa K, Maeda M (2003) Rapid aggregation of gold nanoparticles induced by non-cross-linking DNA hybridization. J Am Chem Soc 125:8102–8103. doi:10.1021/ja034876s
Jie GF, Yuan JX (2012) Novel magnetic Fe3O4@CdSe composite quantum dot-based Electrochemiluminescence detection of thrombin by a multiple DNA cycle amplification strategy. Anal Chem 84:2811–2817. doi:10.1021/ac203261x
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51273084, 51473067). Excellent Youth Foundation of Shandong Provincial 264 (ZR2015JL019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 123 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Zhang, L., Shen, L. et al. Electrochemiluminescence DNA biosensor based on the use of gold nanoparticle modified graphite-like carbon nitride. Microchim Acta 184, 2587–2596 (2017). https://doi.org/10.1007/s00604-017-2234-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-017-2234-z