
Abstract
Decarbonisation of the energy system requires new uses of porous subsurface reservoirs, where hot porous reservoirs can be utilised as sustainable sources of heat and electricity, while depleted ones can be employed to temporary store energy or permanently store waste. However, fluid injection induces a poro-elastic response of the reservoir rock, as well as a chemical response that is not well understood. We conducted uniaxial stress-cycling experiments on quartz sand aggregates to investigate the effect of pore fluid chemistry on short-term compaction (stresses of 0.3–35 MPa, five or ten cycles). Two of the tested environments, low-vacuum (dry) and n-decane, were devoid of water, and the other environments included distilled water and five aqueous solutions with dissolved HCl and NaOH in various concentrations, covering pH values in the range 1–14. In the first loading–unloading stress-cycle, 28–64% of the compaction was inelastic, where fluid-saturated samples produced more compaction than dry samples. In addition, compaction was strongly enhanced in alkaline environments and inhibited in acidic ones, compared to distilled water. With prolonged stress-cycling (up to ten cycles), fluid-enhanced compaction effects disappeared. Acoustic emission data and microstructural analyses revealed that microcracking was prevalent in all samples. We inferred that crack growth was aided by Si–O bond hydrolysis and pH-dependent fluid–solid surface interactions. In addition, crack growth rates reduced with prolonged stress-cycling, leading to less fluid-enhanced compaction with increasing cycle number. Our results imply that fluid injection into a clean, quartz-rich, porous reservoir could evoke or inhibit apparent time-independent inelastic deformation depending on the type of fluid injected.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
To mitigate climate change, efforts are being made to decarbonise our energy system and transition towards low- or zero-carbon energy sources, such as renewable energy in the form of wind, solar or water power (Moriarty and Honnery 2016; Hauer et al. 2018; Kabir et al. 2018; DeCastro et al. 2019), and geothermal energy (Shortall et al. 2015; Limberger et al. 2018). Renewable energy can be temporarily stored in geological formations (Mahlia et al. 2014) in the form of synthetic fuel (Graves et al. 2011), compressed air (Budt et al. 2016), or hydrogen (Tarkowski 2019). Other forms of subsurface storage that have an important role to play during the energy transition include seasonal storage of low-carbon natural gas (Fang et al. 2016; Juez-Larré et al. 2016), short- and long-term storage of captured CO2 (IPCC 2005; Jiang et al. 2010; Underschultz et al. 2011) and permanent disposal of wastewater used in gas production (Lei et al. 2013; Keranen et al. 2014). For these reasons, porous subsurface reservoirs, such as aquifers and depleted hydrocarbon reservoirs, have been receiving more and more attention in recent years, as host rocks for injection of all of the above fluids (Liu and Ramirez 2017; Schimmel et al. 2019b). Injection of a foreign, pressurised fluid, and especially cyclic injection and production, will not only bring about changes to the in situ stress state, but also in the (local) temperature field and chemical environment. On both the short and long term, these changes may influence the mechanical and transport behaviour of the host and seal formations, and hence the performance of the subsurface storage complex.
Injection of a pressurised fluid into a porous reservoir will have a direct effect on the state of stress. An increase in pore pressure (\({P}_{p}\)) relative to the overburden stress (\({\sigma }_{v}\)) leads to a decrease in the effective vertical stress acting on the reservoir rock, as given by Terzaghi’s effective stress principle \({\sigma }_{v, eff}={\sigma }_{v}-{P}_{p}\). This may induce a predictable, poro-elastic response, resulting in the expansion of the reservoir rock (Wang 2000). Furthermore, alteration of the chemical environment may result in fluid-rock interactions, potentially impacting inelastic grain-scale deformation processes affected by pore fluid chemistry, such as dissolution (Hajash et al. 1998; Dove 1999), microcracking (Atkinson and Meredith 1981; Brantut et al. 2012; Brzesowsky et al. 2014a; Schimmel et al. 2019a) or pressure solution (Rutter 1983; Lehner 1990; Schutjens 1991; Dewers and Hajash 1995; Spiers et al. 2004; Gratier et al. 2009). Changes in the rate of these processes could affect the mechanical behaviour of the reservoir rock. An increase in the rate of, for example, microcracking could lead to compaction of the reservoir. Induced reservoir compaction can be the source of unwanted effects, such as induced seismicity and surface subsidence (Nagel 2001; Doornhof et al. 2006; Suckale 2009). Injection/production cycles may have similar effects, as well as potentially producing rock damage phenomena that may lead to changes in reservoir behaviour (Heap et al. 2010). However, the chemo-mechanical impact of fluid injection and production strongly depends on pore fluid chemistry and on the mineralogy of the grains and cement that form the reservoir rock. This means that the effects of fluid-rock interactions on reservoir response are much more difficult to predict than the direct poro-elastic response to fluid injection or production.
Depleted hydrocarbon reservoir sands and sandstones, as well as sandstone aquifers, form key targets for CO2 storage (e.g.,Gibson-Poole et al. 2008; Heinemann et al. 2012; Mao et al. 2014), wastewater storage (Andričević et al. 2009) and geothermal energy production (Moeck 2014). Sand aggregates and sandstones have first-order similarities in mechanical behaviour, such as similar grain-scale pore fluid-rock interactions occurring in both materials. In addition, sandstone behaviour is generally approximated as grains held together by cement bonds (e.g., see Potyondy and Cundall 2004; Einav 2007), implying that sandstones behave as loose granular sand once sufficient cement bonds are broken (Bernabé et al. 1992). This makes sand a suitable analogue to study the behaviour of sandstone.
From compaction experiments on granular quartz, it is well known that the compaction behaviour is affected by the chemical environment. Water-bearing sand samples compact more easily upon increased loading compared to their dry counterparts (Hangx et al. 2010; Brzesowsky et al. 2014a). Similarly, time-dependent (creep) compaction of quartz sand is enhanced in wet versus dry samples, and especially in the presence of alkaline fluids, whereas it is inhibited in acidic environments (Hangx et al. 2010; Schimmel et al. 2019a). These effects of fluid composition on creep are caused by surface interactions between the fluid and the rock-forming minerals, impacting the rate of stress corrosion cracking of grains and grain contacts. However, during injection cycling of the type characterising seasonal storage of energy or geothermal energy production, instantaneous or short-term effects may be more important than long-term creep phenomena, because of the relatively rapid (cyclic) changes in the state of stress. The short-term (i.e., instantaneous or time-independent) compaction behaviour of quartz sand has been investigated in numerous studies employing single-stage loading experiments (Hagerty et al. 1993; Yamamuro et al. 1996; Nakata et al. 2001a, b; Chuhan et al. 2002, 2003; Mesri and Vardhanabhuti 2009; DeJong and Christoph 2009; Wang et al. 2011) and stress-cycling experiments (Monismith et al. 1975; Sawicki and Swidzinski 1995; Wichtmann et al. 2005; Brzesowsky et al. 2014b; Chong and Santamarina 2016; Park and Santamarina 2018). However, in most of these studies, the effect of chemical environment was not systematically studied and/or the stress range investigated was too low to be representative of typical shallow crustal conditions (i.e., up to 100 MPa or 5 km depth), falling below the stress level typically required for significant grain breakage (Zhang et al. 1990; Brzesowsky et al. 2011).
The present study investigates the effect of pore fluid chemistry on the short-term compaction behaviour of quartz sand aggregates. The aggregates are treated as analogues for clean, highly porous, quartz-rich reservoir sands and sandstone. Axial stress-cycling (oedometer) experiments reaching up to 35 MPa were performed on sand at 80 °C, simulating typical reservoir conditions at depths of 2–4 km. Loading rates of 5 MPa/min were used, allowing for discrimination between different acoustic emission (AE) events. Though this loading rate is several orders of magnitude faster than would be employed during injection and/or production operations, it should be noted that for time-independent compaction, loading rate does not seem to significantly affect the compaction behaviour (Brzesowsky et al. 2014b). We systematically tested the effect of chemical environment, including a low-vacuum (dry) control environment, a chemically inert pure fluid (n-decane), and HCl and NaOH pure fluid solutions covering a pH range of 1 to 14. These environments represent chemical conditions relevant to subsurface activities related to geothermal energy production, CO2 or wastewater storage or seasonal energy storage. Insight into the deformation mechanisms operating at the grain-scale was obtained via acoustic emission counting, and by means of microstructural study and grain size analysis applied before and after individual cyclic compaction tests. The tests revealed that compaction was for a large part inelastic and that this permanent compaction was controlled by microcracking, which was in turn strongly influenced by pore fluid chemistry. Wetting with aqueous solutions produced more strain compared to conditions devoid of water. Furthermore, permanent compaction was enhanced in samples saturated with alkaline solutions and inhibited in acidic solutions compared to distilled water-saturated samples. These results imply that, in case of injection of an alkaline solution, the initial stage of a cyclic injection/production operation has the potential to induce enhanced reservoir compaction.
2 Experimental Method
2.1 Sample and Pore Fluid Preparation
The quartz sand used in this study was collected from the Heksenberg Formation at the Beaujean Quarry in Heerlen, the Netherlands. Sample material was prepared as described in previous work (Brzesowsky et al. 2011, 2014a; b) and is the same material used by Schimmel et al. (2019a). The sand was sieved to obtain a grain size fraction of 196 ± 16 µm. X-ray diffraction analysis and inductively coupled plasma emission spectroscopy (ICP-ES) measurements on the sieved material indicated a quartz content of > 99 wt% with zircon and Na-rich feldspar present as principal trace minerals. The feldspar was successfully removed by washing in distilled water using a gravitational separation technique. The present 1D compaction experiments were conducted on 5.01 g samples of the final Beaujean sand material.
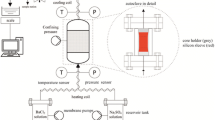
In total, a low-vacuum (dry) control environment and seven pore fluids were employed to investigate the effect of chemical environment on the short-term cyclic compaction behaviour of quartz sand (Table 1). These pore fluids included a chemically inert fluid (n-decane) with respect to quartz, distilled water, three HCl solutions (pH 1, 2.4 and 4) and two NaOH solutions (pH 9 and 14). Low-vacuum (dry) conditions were obtained by evacuating the sample before and during mechanical testing, using a single-stage rotary vacuum pump (Fig. 1a). The n-decane used was commercially available analytical reagent grade. The acid solutions of pH 1, pH 2.4 and pH 4 were prepared by diluting fixed quantities of hydrochloric acid (HCl) in distilled water. The alkaline solutions of pH 9 and pH 14 were prepared by diluting a fixed quantity of sodium hydroxide (NaOH) in distilled water. All aqueous solutions were thoroughly stirred and left to equilibrate for at least 5 days prior to use. After equilibration, each solution was heated to 80 °C and the solution pH determined at the experimental temperature using a portable pH meter that accounts for the temperature-dependence of pH. Note that the solutions were not saturated with respect to quartz.

Overview of the experimental set-up used in this study. a Pore fluid system with Drechsel bottle and vacuum pump. b Instron servo-controlled loading frame employed with a 100 kN load cell and c compaction vessel
2.2 Experimental Set-Up and Acoustic Emission Monitoring System
The stress-cycling experiments were performed in a uniaxial (1D) compaction vessel (oedometer), loaded using an Instron 8862 servo-controlled loading frame (Fig. 1b and c). The compaction vessel is modified from the vessel originally described by Schutjens (1991), and later by other workers (Hangx et al. 2010; Brzesowsky et al. 2014a, b; Schimmel et al. 2019a). The vessel and loading pistons are constructed from corrosion resistant Monel K-500, a copper-nickel-molybdenum alloy. The bottom piston and vessel are secured on the loading frame. The top piston contains a pore fluid bore, allowing both evacuation of the sample and injection of a pore fluid. The sand sample fills the bore of the vessel (20 mm diameter) between the upper and lower pistons, having an initial height of approximately 10 mm. A 1 mm thick, porous, stainless steel plate between the top piston and sample ensures equal distribution of fluid over the sample cross-sectional area upon injection. In addition, it prevents grains from entering and clogging the pore fluid bore. Both the top and bottom piston are sealed against the vessel wall using EPDM O-rings. Sample evacuation and fluid introduction are achieved via the pore fluid system, consisting of a vacuum pump and a Drechsel bottle (Fig. 1a). For evacuation of the sample, the vacuum pump is connected to the sample via the Drechsel bottle. In addition, the Drechsel bottle is used for flooding the sample with pore fluids.
Axial force, displacement and temperature are controlled and measured throughout the experiment. Force is applied by advancing the Instron loading ram. Force is measured externally using the Instron load cell (0–100 kN range, resolution ± 0.05 kN) and using an internal load cell (0–100 kN range, resolution ± 0.05 kN) located in the top part of the bottom piston. Piston position and displacement are measured using linear variable differential transformers (LVDT) located in the Instron drive unit (± 50 mm range, resolution ± 0.25 µm) and a Sangamo LVDT (± 1 mm range, resolution ± 0.1 µm) located between the upper piston and the vessel (Fig. 1c). A furnace allows controlled heating of the sample with ± 0.5 °C accuracy using a K-type chromel–alumel control thermocouple. This thermocouple is positioned within the furnace windings and connected to a proportional-integral-derivative (PID) controller. A second K-type thermocouple, embedded in the vessel wall adjacent to the sample, independently measures sample temperature.
In addition, the experimental set-up is equipped with an acoustic emission (AE) monitoring system that detects and counts AEs produced by the compacting sample. The AE events are detected using a ceramic piezoelectric resonator, mounted externally on the top piston (Fig. 1c). The resulting signal passes through a precision preamplifier (36 dB gain) and a multistage signal conditioning system (24 dB gain). A 100 kHz to 1 MHz band-pass filter is applied to eliminate low frequency interference and sensor resonance effects. Lastly, a two-counter channel with a constant trigger-threshold of 200 mV, which is just above the noise level, discriminates and continuously counts the incoming AE events. Pulse stretching times (PST) of 1000 µs and 500 µs are set for each counter, to check for wave pocket arrival-bouncing effects and counter saturation. In case of discrete events, the count rates from the two channels should be identical.
2.3 Experimental Procedure
Vessel assembly was conducted outside the Instron frame, first mounting the compaction vessel on its lower piston. A 10 mm high stainless steel spacer was then inserted into the vessel, to position the sample in the middle of the vessel. Subsequently, a Teflon liner was emplaced to reduce friction between the sand sample and vessel wall. This liner is 10 mm high and 50 μm thick, and rims the inside of the vessel. Before emplacing the liner, the Teflon was sprayed with Molykote D-312R and dried for at least 1 hour to further reduce friction. The sample material was then funnelled into the vessel and the porous steel plate was carefully placed on top of the sample using a guiding rod. The furnace and top piston were subsequently added and the entire assembly located into the Instron loading frame.
In preparation for each experiment, the sample was heated to the target temperature of 80 °C, while simultaneously evacuating the sample assembly. After stabilisation at 80 °C, which took approximately 1.5 h, each sample was pre-compacted by stress-cycling, while continuously evacuating (Stage I, Fig. 2). First, a small stress of 0.3 MPa (equivalent to a force of 0.1 kN) was applied to the sample. The applied stress was then cycled at a constant stress rate of 5 MPa/min following the sequence shown in Fig. 2 (i.e., 0.3 – 5 – 0.3 – 15 – 0.3 – 25 – 0.3 – 35 – 0.3 MPa). The start of Stage I stress-cycling (tI = 0) is defined as the moment this loading sequence was initiated (Fig. 2). The aim of Stage I stress-cycling was to condition each sample such that the variability in porosity between different samples was reduced in preparation for subsequent testing. This was done because compaction of sand aggregates is highly sensitive to the initial microstructure (Hangx et al. 2010; Brzesowsky et al. 2014a).

Experimental procedure employed in this study, consisting of two stages of stress-cycling. Stress-cycling in Stage I is under low-vacuum (dry) conditions for all experiments and commences at tI = 0. Stage II starts at tII = 0 and is either at low-vacuum or fluid-saturated conditions and includes either five (as shown) or ten stress-cycles (not shown). Note that for fluid-saturated experiments, fluid was introduced prior to the start of the first cycle of Stage II, i.e., before tII. The applied stress is the stress measured by the external load cell
After pre-compaction, a second stage of stress-cycling (Stage II) ensued, where the stress was cycled between 0.3 and 35 MPa in five or ten cycles at a constant stress rate of 5 MPa/min (Table 1). The start of Stage II stress-cycling (tII = 0) is defined as the moment the second loading ramp was initiated (Fig. 2). This stage was performed either at evacuated (dry) or fluid-saturated conditions. For low-vacuum (dry) experiments, evacuation continued throughout the experiment duration. For fluid-saturated conditions, the pore fluid was introduced prior to the start of Stage II stress-cycling by vacuum-flooding the sample at 0.3 MPa applied stress (Fig. 2). The pore fluid pressure was maintained at atmospheric pressure by drainage to air, so that all axial stresses quoted above are effective stresses. For each environment, three to four repeat experiments were conducted to ensure reproducibility of the results (Table 1).
After the second stress-cycling stage, the experiment was stopped. The experiment was terminated by completely unloading the sample and switching off the furnace. In case of the low-vacuum (dry) experiments, subsequently the vacuum was removed. The sample was then allowed to cool to room temperature, followed by removal of the assembly from the Instron loading frame. The sample and Teflon liner were, subsequently, gently pushed out of the vessel into a container using a glass piston. Samples tested with pore fluids were wet and cohesive due to capillary forces, so remained intact during extraction. Dry samples were vacuum-flooded with distilled water prior to removal, allowing for intact extraction. As this vacuum-flooding occurred under near-zero applied stress, i.e., the piston was just touching the sample, no effect of flooding on the sample microstructure is expected. All samples, surrounded by their Teflon liner, were subsequently placed in an oven at 50 °C to dry for a minimum of 5 days prior to preparation for microstructural analysis.
2.4 Data Acquisition and Processing
Throughout each experiment, external Instron load, internal load, Instron LVDT position, Sangamo LVDT position, sample temperature and cumulative number of acoustic emission counts were logged at an interval of 1 second. Applied axial stress and effective axial stress were calculated from the external Instron load and internal load data. Friction between the top piston seal and vessel wall was negligible for these experiments (< 0.6 MPa). However, comparison of external and internal load cell data demonstrated significant friction at the sample-vessel interface, despite measures to reduce friction between the sand sample and vessel wall. At the maximum applied stress, the stress measured by the internal load cell varied between 75.4 and 86.0% of the applied stress. The sample stress, i.e., the average of the internally and externally measured stress, therefore reflects the average stress state in each sample. The spread in sample stress between the different samples varied with applied stress, i.e., at the maximum applied stress the sample stress was 31.7 ± 0.7 MPa (± 2.2%), while at 5 MPa applied stress a sample stress of 4.3 ± 0.4 MPa (± 9.3%) was found for all samples.
The length of the sample at the start of Stage I and Stage II stress-cycling, i.e., the sample length at tI or tII (Fig. 2), was determined by comparing the Instron LVDT position to an empty vessel reference point, at the same applied stress of 0.3 MPa. The Sangamo LVDT displacement data was corrected for elastic machine distortion using a predefined, eighth order polynomial determined from machine calibrations on the experimental set-up without a sample. Instantaneous volumetric strain, defined as \({e}_{v}= -\Delta L/{L}_{0}\), was calculated from the corrected Sangamo LVDT displacement data (error ± 0.02%), where \({L}_{0}\) is the length of the sample at the start of Stage I or Stage II stress-cycling and \(\Delta L\) is the change in length at any subsequent instant.
In addition, to investigate the strain response to increasing the applied stress, the sample stiffness per upward stress increment was determined, following Chuhan et al. (2003). The sample stiffness or constrained modulus is defined as \(M=\Delta \sigma /\Delta {e}_{v}\) and was determined for small increments in sample stress \(\Delta \sigma\) and the resulting incremental volumetric strain \(\Delta {e}_{v}\). \(M\) was calculated by performing a least-squares inversion over a variable strain window centred around each individual stress data point. The window size was based on a set tolerance for the strain of 1%, such that the error in the sample stiffness was always ≤ 1.6%.
2.5 Analytical and Microstructural Methods
After the experiments, grain size, microstructural and crack density analyses were performed on selected samples (Table 1). Grain size analysis on 1 g portions of selected samples and on undeformed starting material was performed using a Malvern laser diffraction particle sizer. This allowed determination of the average grain size and grain size distribution before and after deformation. Despite the fact that laser particle size analysis systematically overestimates grain size by approximately 25%, due to fines adhering to coarse grains (Hangx et al. 2010; Pluymakers et al. 2014), the results are preferred here over optical analysis as they avoid stereological uncertainty inherent to 2D section study and sample a larger grain population.
Samples selected for microstructural and crack density analyses were impregnated with a low-viscosity, blue-dyed (Oil Blue organic dye, DuPont), epoxy resin (Araldite 2020) after drying. The blue-dyed resin resulted in a clear distinction between pores and quartz grains. Thin sections were cut parallel to the loading axis and analysed using transmitted light microscopy. Micrograph mosaics (50× magnification) covering the entire thin section, with a typical dimension of 18 by 6 mm, were obtained for crack density analysis. The micrograph of a sample extracted immediately after Stage I covered a slightly smaller area (13.3 by 6.6 mm).
Crack density analysis involved manual tracing and counting of cracks. Partially through-going cracks with clear ingress of blue epoxy were counted. In addition, cracks were included that resulted in the breaking of grains into two or three fragments. These fragments were interpreted to originate from the same grain based on colour, shape and fragment position. Small fragments, resulting from grain shattering or chipping, were excluded from the analysis. This method of crack determination most likely led to an underestimation of the actual number of cracks, but provided a basis to compare crack densities from different samples. Subsequently, crack density, defined as the number of cracks per area (mm−2; error ± 0.01 mm−2), was calculated. In some samples, air bubbles, scratches and other types of damage of the thin section prevented counting of cracks over the entire imaged area. In these cases, the size of the uncountable areas was determined and subtracted from the total area before calculating the crack density. For most samples, an area between 105 and 108 mm2 was analysed. For the selected NaOH pH 14 experiment (14pH-02) and a sample extracted after Stage I, cracks were counted over an area of 79 mm2 and 87 mm2, respectively.
3 Results
3.1 Stage I Pre-Compaction at Low-Vacuum (Dry) Conditions
All samples were pre-compacted in Stage I by means of stress-cycling at low-vacuum (dry) conditions. To illustrate the mechanical and acoustic data of this stage, the data of two representative experiments (i.e., vac-04 and DI-04) are presented in Fig. 3a and b. Compactive strain and cumulative AE count increased with each successive stress-cycle. By the end of pre-compaction, more permanent strain was produced in the high porosity sample (DI-04, φI = 44.0%) compared to the low porosity one (vac-04, φI = 43.6%), i.e., 5.6% strain compared to 4.5% strain. In both samples, approximately 110,000 AEs were measured. A similar strain and AE response to stress-cycling during pre-compaction was observed for the other samples.

Typical results of Stage I pre-compaction at 80 °C and low-vacuum (dry) conditions. a Applied stress versus strain and b cumulative AE count versus strain plotted for two representative experiments, i.e., vac-04 and DI0-4, with starting porosities (φI) of 43.6% and 44.0%, respectively. c Total AE count, and total and permanent strain accumulated during Stage I for all samples. d Porosity reduction during Stage I for all samples. In d, iso-porosity-reduction lines reflect 4%, 3% and 2% absolute change in porosity
For all samples, an overview of the total and permanent strain and cumulative AE count developed during Stage I pre-compaction is presented as a function of porosity prior to pre-compaction in Fig. 3c. For the entire sample suite, permanent strain resulting from pre-compaction varied between 3.1 and 6.2%, reflecting 65–78% of the total strain. Strain was accompanied by AE activity, varying between approximately 90,000 and 112,000 events for all samples, and both strain and AE count increased with sample porosity. The effect of porosity on sample compaction is made more explicit in Fig. 3d. Pre-compaction reduced individual sample porosity by 1.9–3.7% (see contours Fig. 3d), reducing the overall porosity range of the sample suite from 43.0 ± 1.6% to 40.2 ± 1.0%. The high porosity samples thereby had a relatively stronger reduction in porosity than the low porosity samples.
3.2 Stage II Stress-Cycling Behaviour
3.2.1 Aggregate Compaction at Low-Vacuum (Dry) Conditions
Typical results of Stage II stress-cycling are presented in Fig. 4, which shows two experiments conducted at low-vacuum (dry) conditions, i.e., vac-03 and vac-04, that were stress-cycled five and ten times, respectively. In all experiments, strain accumulated during each stress-cycle, which was only partly recovered during unloading, i.e., 57–60% of the total compactive displacement measured after five stress-cycles was inelastic (Fig. 4a and b). Overall, it could be observed that, during stress-increase, the stress–strain curve transitioned from being initially quasi-linear to becoming concave downward near the peak stress (Fig. 4b). With each subsequent cycle the stress–strain curve became more linear, as well as steeper. Unloading after peak stress, resulted in an initially steep (almost vertical) stress–strain curve that transitioned into a gentler slope with decreasing stress. This unloading behaviour was similar for each stress-cycle.

Typical results of Stage II stress-cycling at 80 °C. a Strain versus time, b applied stress versus strain, and c cumulative AE count versus total strain plotted for two low-vacuum (dry) experiments vac-03 (φII = 40.0%) and vac-04 (φII = 40.8%). Vac-04 was stress-cycled ten times. Zero strain and AE reference is at the start of Stage II
Furthermore, a clear effect of sample porosity (i.e., porosity after Stage I pre-compaction) on the amount of compaction could be discerned, as illustrated by the two dry experiments with different sample porosities presented in Fig. 4. In the low porosity sample (vac-03, φII = 40.0%), 0.7% permanent or inelastic strain was measured after the first stress-cycle, while in the high porosity sample (vac-04, φII = 40.8%) 0.9% inelastic compaction was observed. Each successive cycle led to more total strain with systematically more total and inelastic strain developing in the high porosity sample, though the amount of additional permanent strain decreased with each cycle. At the end of the fifth stress-cycle, the cumulative inelastic strain amounted to 2.1 and 2.6% for the low and high porosity sample, respectively. Continued stress-cycling produced 3.7% of inelastic strain after ten cycles.
In all experiments performed, compaction was associated with acoustic emissions, as illustrated in Fig. 4c for the selected samples. During the initial quasi-linear loading phase, relatively few AEs were counted. However, once the stress–strain behaviour deviated from quasi-linearity, a rapid increase in cumulative AE count was observed, i.e., at approximately 2% strain during the first cycle (cf. Figure 4b and c). In each subsequent stress-cycle, the AE count rapidly increased once the maximum amount of strain obtained in the previous cycle was exceeded. Unloading also produced a small number of AEs, especially in the first few cycles. In line with the effect of porosity on strain, in the low porosity sample (vac-03) approximately 27,000 events were counted during the first stress-cycle, compared to approximately 32,000 events in the high porosity sample (vac-04). With each successive cycle the number of newly accumulated AEs decreased. After the fifth stress-cycle, total cumulative AE count increased to approximately 72,000 and 87,000 for the low and high porosity sample, respectively. After the tenth stress-cycle, 113,000 AE events were measured for sample vac-04. Similar trends in strain and AE were shown by all samples tested.
3.2.1.1 3.2.1.1 Total, Elastic and Permanent Strain per Stress-Cycle
Pre-compaction (Stage I) and the data presented in Sect. 3.2.1 showed that compaction behaviour strongly depends on sample porosity (cf. Sect. 3.1), as also demonstrated by previous studies (Nakata et al. 2001b; Chuhan et al. 2003; Brzesowsky et al. 2014b). Therefore, to exclude porosity effects, the Stage II results are grouped according to: Group A with porosities in the range 39.9–40.3%, and Group B with porosities in the range 40.4–40.8% (see Table 1). Note that for each group, all chemical environments are represented, with the exception of the distilled water environment in Group A. In addition, stress-cycling of two samples in Group B (vac-04 and DI-04) continued up to ten cycles.
From the unloading data obtained during Stage II runs, it was possible to resolve the amount of inelastic compaction, allowing the plotting of total, elastic and permanent strain per stress-cycle as done in Fig. 5. In this case, the zero reference is at the start of each stress-cycle. In all experiments, the three strain types decreased with successive cycling, i.e., less additional strain was accumulated with each subsequent stress-cycle. In addition, the high porosity samples generally produced more total, elastic and permanent strain. In the low-vacuum (dry) environment, 2.5–2.8% total strain was accumulated in the first stress-cycle, which reduced to 1.9–2.0% total strain by the fifth cycle (Fig. 5a, b). By contrast, the amount of elastic strain per stress-cycle decreased only slightly from 1.8 to 1.6–1.7% (Fig. 5c, d). Furthermore, over the course of the five stress-cycles, the contribution of permanent strain decreased from 0.7–0.9% in the first stress-cycle to ~ 0.3% in the fifth stress-cycle (Fig. 5e, f). Continued stress-cycling further reduced the strain accumulated per cycle, i.e., 1.7, 1.5 and ~ 0.3% total, elastic and permanent strain was measured in the tenth cycle, respectively.

a, b Total strain, c, d elastic strain, e, f permanent strain and g, h cumulative AE count per stress-cycle (i.e., zero strain and AE reference at the start of each stress-cycle) versus pH for porosity Group A (39.9–40.3%) and B (40.4–40.8%). Note the difference in scale between total, elastic and permanent strain. In e, f and g, h, the total permanent strain and total cumulative AE count over the five stress-cycles with respect to the start of Stage II is added, respectively
3.2.1.2 AE Activity
Similar to the trends observed in strain, cumulative AE count per stress-cycle significantly decreased with increasing cycle number for all experiments. In addition, the AE count was overall higher in the more porous sample (vac-04). In the first stress-cycle, 27,000–32,000 AEs were measured in low-vacuum (dry) conditions, which reduced to 7,000–8,000 counts per cycle in the fifth stress-cycle (Fig. 5g, h). In the tenth stress-cycle, only 4,000 AEs were measured. In Fig. 6, the cumulative AE count is plotted as a function of cumulative permanent strain (i.e., with respect to the start of Stage II), showing the increase of these quantities per stress-cycle. For both the low and high porosity sample, the data can be approximated by a slope 32,000 AEs/% permanent strain for the first five stress-cycles. At higher stress-cycles, the slope becomes less steep (Fig. 6b).

a, b Cumulative AE count versus cumulative permanent strain for porosity Group A (39.9–40.3%) and B (40.4–40.8%)
3.2.1.3 Aggregate Stiffness
The sample stiffness or constrained modulus (\(M\)) was not constant during loading (Fig. 7a, b). During loading in the first stress-cycle, \(M\) steadily increased from 0.6 to 0.7 GPa for low-vacuum (dry) samples, with an inflection point around 10 MPa, until reaching a peak value of 1.6–1.7 GPa at approximately 15 MPa sample stress. After attaining the peak value, \(M\) decreased with increasing stress, to a value slightly higher than the stiffness at the start of the cycle (1.0–1.1 GPa). With each subsequent cycle, the initial stiffness, i.e., at near-zero applied stress, increased only slightly. However, the peak and final stiffness increased significantly with more cycles, to a peak \(M\)-value of 2.1–2.2 GPa and a final \(M\)-value of 1.6–1.9 GPa in the fifth cycle. Furthermore, as the number of cycles increased, \(M\) remained approximately constant upon reaching the peak value, before decreasing again. By the end of the fifth cycle, the stress range over which \(M\) remained near-constant spanned roughly 15–26 MPa. In the tenth stress-cycle, peak stiffness had increased to 2.4 GPa (Fig. 7b). Interestingly, AEs were not measured directly at the start of loading, but started to accumulate after approximately 10 MPa sample stress in the first stress-cycle and significantly increased after approximately 15 MPa sample stress (Fig. 7c, d). With repeated stress-cycling, the initiation of AE activity shifted to higher stresses.

a, b Sample stiffness or constrained modulus (\(M\)) versus sample stress for the low-vacuum (dry) and 4 pH HCl solution experiment in Group A, and low-vacuum (dry) and distilled water experiment in Group B. Only the first and fifth, and, in case of Group B, the tenth cycle are shown. c, d Cumulative AE count during loading versus sample stress for the low-vacuum (dry) and 4 pH HCl solution experiments in Group A, and low-vacuum (dry) and distilled water experiment in group B. Only the first and fifth, and, in case of Group B, the tenth cycle are shown
3.2.2 The Effect of Chemical Environment on Aggregate Compaction
The samples saturated with fluids showed qualitatively the same compaction behaviour as the low-vacuum (dry) samples described in Sect. 3.2.1. However, quantitatively there were some marked differences, which can be attributed to the chemical environment. Samples saturated with fluids produced more total and permanent strain than their dry counterparts of similar porosity. This effect was most pronounced in the first stress-cycle (Fig. 5a, b and e, f). Flooding with the most alkaline fluid (pH 14 NaOH solution) produced the largest increase in strain, i.e., 4.0–4.2% total strain (Fig. 5a, b) of which 2.5–2.7% was permanent (Fig. 5e, f). By contrast, non-aqueous (n-decane) or highly acidic (pH 1 or pH 2.4 HCl solution) environments showed only 2.8–3.0% total strain, of which 0.9–1.2% was permanent. The chemical environments with intermediate pH values showed permanent strain values that clearly fall within these two extremes, suggesting that permanent strain roughly relates to fluid pH, increasing with increasing pH. With each subsequent stress-cycle the spread in total and permanent strain initially observed between the different chemical environments diminished. Though in the fifth stress-cycle the fluid-rich environments still induced more total and permanent compaction compared to low-vacuum (dry) conditions, the effect of individual fluids was nearly indiscernible.
Contrary, the elastic strain accumulated per stress-cycle appeared to be relatively insensitive to chemical environment (Fig. 5c, d). The largest elastic strains were measured in samples saturated with n-decane (approximately 1.9% in the first cycle), while the smallest elastic strains were measured in pH 14 NaOH-saturated samples (approximately 1.5% in the first cycle). For the other chemical environments, elastic strain values between these outer bounds were observed, showing no clear dependence with respect to fluid composition. This led to the range of elastic strains being 1.4–1.7% by the end of the fifth cycle. Furthermore, elastic strain appeared to be near-independent of sample porosity, as the two porosity groups yielded similar elastic strain values (c.f. Fig. 5c and d).
3.2.2.1 AE Activity
For most fluids, fluid-saturated samples produced more AEs than dry samples, which was again most pronounced in the first stress-cycle. Exceptions were the samples saturated with an inert pore fluid (n-decane; 28,000–29,000 AEs) and highly alkaline solution (pH 14 NaOH solution; 32,000–37,000 AEs), producing less AEs than the dry samples (Fig. 5g, h). The samples saturated with the remaining fluids, i.e., fluids in the pH range 1–9, produced approximately 54,000–63,000 AEs, with no clear dependency on fluid pH. With each subsequent stress-cycle, the number of AEs counted decreased, as did the difference between the various fluids. Plotting cumulative AE count versus cumulative permanent strain (i.e., with respect to the start of Stage II) revealed that significantly more AEs were produced per permanent strain increase in the samples saturated with aqueous solutions in the pH range 1–9, i.e., between 36,000 and 53,000 AEs per percent permanent strain, than in the dry samples that were characterised by a slope of 32,000 AEs/% permanent strain (Fig. 6). Notably, even smaller slopes were observed in the n-decane-saturated samples and the samples saturated with the most alkaline fluid (pH 14 NaOH solution), i.e., slopes of 18,000 and 5,000–14,000 AEs/% permanent strain, respectively. Similar to observations for the dry samples, in the fluid-saturated samples AE activity in the first stress-cycle initiated around 10 MPa sample stress and significantly increased around 15 MPa stress. The onset of AE activity shifted to higher stresses with successive cycling (Fig. 7c, d).
3.2.2.2 Aggregate Stiffness
Qualitatively similar trends in aggregate stiffness were found in the fluid-saturated samples compared to the dry ones. However, quantitatively some differences were observed (Fig. 7a, b). Overall, \(M\) was lower and, consequently, the peak stiffness was lower for the fluid-saturated samples compared to the dry ones. With increased stress-cycling the difference in \(M\) became smaller for the different chemical environments and eventually vanished in the tenth stress-cycle. The peak stiffness values of the samples saturated with pH 4 HCl solution and distilled water were 1.2–1.4 GPa in the first stress-cycle, increasing to 2.0–2.1 GPa during the fifth cycle. In the tenth stress-cycle, \(M\) of the water-wet sample was slightly higher than the dry sample, with a nearly indiscernible peak stiffness of 2.5 GPa.
3.3 Microstructural Observations
3.3.1 General Observations
The undeformed material consisted of sub-rounded grains with a narrow grain size distribution and a mean grain size (D50) of 245 μm (Fig. 8a). As a baseline, a thin section of a sample subjected to only Stage I pre-compaction was prepared for crack density analysis, which yielded a crack density of 0.94 mm−2 (Fig. 8b). In all samples subjected to Stage II stress-cycling, the overall grain size decreased, producing fine material (< 100 μm), and the crack density increased (Fig. 8). Numerous cracks, which were evenly distributed throughout the sample, were observed in all samples. Intragranular cracks were seen within grains, resulting in grain failure (Fig. 9). Furthermore, interference of cracks was observed (see Fig. 9a, c and d), as well as multiple cracks emerging from the same grain-to-grain contact area (Fig. 9c and d). Some chipped grains were distinguished. Small grain fragments were prevalent in all samples, which commonly clustered around grain-to-grain contacts (dashed ellipses in Fig. 9b and d), suggesting contact crushing, though no attempts were made to quantify the number of cracks in these contact regions.

a Grain size distribution for selected samples and undeformed material. The inset in a shows the mean grain size (D50) values for different chemical environments. b Mean grain size and crack density versus pH of selected samples, including an undeformed sample and a sample that was only subjected to Stage I stress-cycling. Solid symbols represent mean grain size (left axis) and open symbols represent crack density (right axis)

Microstructures of a selection of samples spanning the whole of the tested pH spectrum, including a low-vacuum (dry) conditions, b n-decane fluid, c distilled water and d pH 9 NaOH solution. Cracks are indicated by arrows, where “ti” and “pi” indicate through-going and partial intragranular cracks, respectively. Dashed ellipses indicate areas with grain-to-grain contact crushing. The loading direction is vertical
It should be noted that all samples remained cohesionless, even after ten stress-cycles, with the exception of samples saturated with pH 14 NaOH solution. Upon retrieval, these samples were slightly cohesive and the grains appeared lightly cemented, though cementation was not evident from the thin section and no dissolution–precipitation features could be recognised.
3.3.2 Effects of Chemical Environment on Grain Size and Crack Density
Stress-cycling at low-vacuum (dry) conditions resulted in a decreased mean grain size of 227 μm (i.e., a 7% decrease in mean grain size; Fig. 8), along with an increase in crack density to 1.52 mm−2 (Fig. 8b). As also suggested by the AE data, the presence of fluids led to a further decrease in mean grain size and a concomitant increase in crack density (Fig. 8). Compared to dry compacted material, grain size reduction in the inert fluid environment (n-decane) amounted to a 10% decrease in mean grain size (D50-value of 221 μm), and resulted in a crack density of 1.74 mm−2. The presence of aqueous fluids led to more grain size reduction and increased crack densities. The largest grain size reduction (13% reduction; D50 = 212 μm) and increase in crack density (2.22 mm−2) were observed in the pH 14 NaOH solution experiment. The degree of grain size reduction diminished with decreasing solution pH, i.e., 13, 12, 10 and 11% grain size reduction was measured in the samples saturated with distilled water, pH 4 HCl solution, pH 2.4 HCl solution and pH 1 HCl solution, respectively (see inset Fig. 8a and b). However, the D50 of 218 μm (11% reduction) determined from the sample flooded with pH 9 NaOH solution fell outside this pH trend. The experiments conducted with low pH HCl solutions, pH 2.4 and pH 4, were characterised by relatively low crack densities of 1.59 mm−2 and 1.55 mm−2, respectively (Fig. 8b). By contrast, the pH 1 HCl, distilled water and pH 9 NaOH solution experiments had higher crack densities of 2.08 mm−2, 2.08 mm−2 and 2.06 mm−2, respectively. Overall, the samples saturated with aqueous solutions showed a minimum in grain size reduction and crack density at pH 2.4, increasing with higher/lower solution pH. Hence, the microstructural observations agree well with total permanent strain accumulated during each experiment (cf. Figs. 5e, f and 8b).
4 Discussion
The present Stage I and Stage II stress-cycling experiments on quartz sand up to 35 MPa showed that compaction strongly depends on initial sample porosity, but even more so on chemical environment. Wetting of quartz sand resulted in more compaction and a higher AE count compared to stress-cycling at low-vacuum (dry) conditions, with more pronounced effects in the presence of aqueous solutions, compared to inert fluid. Moreover, alkaline solutions appeared to induce more compaction than samples compacted in the presence of acidic solutions. However, this pH effect was most pronounced in the first stress-cycle and diminished with each subsequent cycle. During the first stress-cycle, 28–64% of the measured compaction was permanent, which decreased to 16–25% by the fifth stress-cycle. The amount of elastic deformation only slightly decreased during stress-cycling. The progressive compaction of the sand aggregates resulted in an increase in peak sample stiffness over multiple stress-cycles. In addition, during loading, the sample stiffness increased to a peak value at 15 MPa sample stress and reduced again in the first stress-cycle, but remain constant after reaching the peak value in later stress-cycles. Grain size and crack density analysis clearly indicated that grain breakage resulted in grain size reduction. Moreover, grain size reduction and crack density were more pronounced in fluid-saturated samples compared to dry ones, and intensified with increasing solution pH.
In the following, we will first discuss the processes controlling deformation during cyclic loading, which includes a discussion on the sample stiffness during loading. Then, we will summarise the theory of crack growth and the effect of aqueous fluids on crack growth, as a basis for interpreting the effects of chemical environment on compactive strain observed in our experiments. Lastly, the implications of our observations for several cyclic injection scenarios are discussed.
4.1 Porosity Reduction during Stage I Stress-Cycling at Low-Vacuum (Dry) Conditions
Stage I pre-compaction stress-cycling reduced sample porosity via, presumably, intergranular sliding, grain rotation and rearrangement (Takei et al. 2001; Chuhan et al. 2003; Mesri and Vardhanabhuti 2009; Omidvar et al. 2012), and to some extent grain breakage, as evidenced by the AE data (Fig. 3b and c). Slightly more deformation developed in higher porosity samples compared to lower porosity ones (Fig. 3). This was likely caused by two factors. Firstly, in the higher porosity samples, slightly more space was available to accommodate grain rearrangement before grain breakage (Hagerty et al. 1993; Brzesowsky et al. 2014b). Secondly, in the higher porosity samples, on average the coordination number was probably slightly lower compared to the lower porosity samples, resulting in higher grain contact stresses, thereby promoting grain crushing/breakage, as evidenced by the AE data (Fig. 3b and c; Brzesowsky et al. 2014b). Most of the initial high porosity samples retained an overall higher porosity after pre-compaction (Fig. 3d). This is typically also observed in soil mechanics studies (Hagerty et al. 1993; Nakata et al. 2001b). However, Stage I pre-compaction revealed that initially high porosity samples produce a relatively larger change in sample porosity than low porosity samples (Fig. 3d).This suggests that the amount of pre-compaction strain does not directly depend on the starting porosity, which makes it difficult to predict sample porosity after pre-compaction on the basis of starting porosity alone. This underlines the importance of having samples of similar porosity in compaction experiments employing loose granular aggregates.
4.2 Sample Stiffness during Loading in Stage II Stress-Cycling
In each stress-cycle of Stage II, stiffening of the samples was observed during the initial part of loading (applied stress < 15 MPa, Fig. 7a, b). After reaching a peak value, \(M\) decreased in the first stress-cycle, while in the later cycles (e.g., cycles 5 and 10), \(M\) remained constant up to approximately 26 MPa applied stress (Fig. 7a, b). Stiffening of the sample is classically inferred to be caused by closer packing of the grains via particle rearrangement (Chuhan et al. 2003), resulting in a locked aggregate state (Mesri and Vardhanabhuti 2009). The so-called unlocking mechanisms include grain damage, grain slip and grain rotation, which allow further compaction of the aggregate, while reducing the stiffness of the sample (Mesri and Vardhanabhuti 2009). The change in \(M\) with increasing applied stress can therefore be used to evaluate whether locking, i.e., increasing \(M\), or unlocking, i.e., decreasing \(M\), mechanisms dominate compaction.
During loading in the first stress-cycle, \(M\) increased approximately linearly with increasing applied stress, though an inflection point around 10 MPa stress was observed (Fig. 7a, b). This stress coincides with the initiation of AE activity in the first stress-cycle (Fig. 7c, d), suggesting that grain damage initiated. However, as \(M\) continued to increase with loading, the contribution of grain damage was insufficient to significantly unlock the sample at this stage. At these low stresses, grain damage likely involved surface grinding and breakage of asperities and, perhaps, some breakage of surface protrusions and sharp particle corners, as grain splitting into one or multiple fragments usually occurs at higher stresses and would produce a stronger unlocking effect (Nakata et al. 2001a; Takei et al. 2001; Chuhan et al. 2002, 2003; Mesri and Vardhanabhuti 2009—see also Fig. 9). Similar inflection points at 10 MPa sample stress were observed in the fifth and tenth cycle (Fig. 7a, b), suggesting that also in these stress-cycles grain damage initiated at this point.
With further loading in the first stress-cycle, \(M\) reached a peak value at approximately 15 MPa sample stress and decreased with increasing stress (Fig. 7a, b). The peak and subsequent decrease in \(M\) was accompanied by strong AE activity (Fig. 7c, d), suggesting that significant grain breakage occurred, such that the unlocking mechanisms dominated deformation, reducing the sample stiffness. As the number of stress-cycles increased to 5 and 10, \(M\) remained nearly constant after reaching the peak value, i.e., a fully locked aggregate, the onset of AE activity occurred at much higher applied stress (approximately 26 MPa; Fig. 7), which coincided with a decrease in \(M\), indicating that unlocking effects were dominating again (Mesri and Vardhanabhuti 2009).
4.3 Mechanisms Controlling Compaction during Stage II Stress-Cycling
In Stage II stress-cycling, 36–72% of the compaction was elastic in the first stress-cycle (Figs. 4 and 5), presumably caused by elastic deformation of the grains and grain contacts. Deformation was accompanied by AE activity (Figs. 4 and 5) and an increase in crack density and grain size reduction (Figs. 8 and 9), suggesting that permanent compaction was caused by particle breaking and crushing, followed by rearrangement of fragments and redistribution of interparticle stresses (Takei et al. 2001; Brzesowsky et al. 2014b).
4.3.1 Crack Growth Leading to Grain Breakage
Grain breakage, through crack propagation, occurs when the tensile stress at the tip of a pre-existing flaw or crack overcomes the tensile strength of the material (Griffith 1920). Previous studies have shown that the average tensile stress in a particle depends on the applied macroscopic stress and the coordination number of the particle, which controls the induced tensile stresses (Jaeger 1967; McDowell and Bolton 1998). At constant macroscopic stress, the induced tensile stress in a particle increases with decreasing coordination number, as the contact stresses increase. In addition, the tensile strength of a grain scales inversely with grain size (Lade et al. 1996; McDowell and Bolton 1998; Nakata et al. 2001b), which is typically explained by the increase in size of Griffith-type flaws, which are assumed to increase with grain size (Zhang et al. 1990). Under stress conditions conducive to crack growth, these flaws can propagate and lead to grain failure.
In a pack of spherical particles in point contact, the largest tensile stress is expected at the edge of the grain-to-grain contact (Hertzian contact theory; Hertz 1882), leading to the development of cracks from the grain contact area, as is often observed (Fig. 9). During crack propagation, strain energy is released. When the strain energy release rate \(G\) equals a critical value, equilibrium or critical crack growth occurs (Lawn 1975; Rice 1978). This critical value is given as \({G}_{c}=2\gamma\), where \(\gamma\) is the surface energy per unit area [J m−2], and is also known as the fracture energy. Additionally, an equivalent failure criterion can be based on the stress field around the crack tip, which can be described through the stress intensity factor \({K}_{I}\) (Irwin 1958). For a small, mode I (opening mode) crack \({K}_{I}=Y{\sigma }_{r}{\left(\pi c\right)}^{1/2}\), where \(Y\) is a dimensionless factor equal to 1.12 for an edge crack, \({\sigma }_{r}\) is the remotely applied stress [MPa] and \(c\) is the half-length of the flaw or crack [μm] (Atkinson 1984; Zhang et al. 1990; Lawn 1993). Equilibrium or critical crack growth will occur when the stress intensity factor equals the fracture toughness, i.e., \({K}_{I}={K}_{IC}\). The fracture toughness depends on the material’s Young’s modulus (\(E\)), surface energy and crack half-length according to \({K}_{IC}={\left(2E\gamma /\pi c\right)}^{1/2}\) (Griffith 1920; Orowan 1944). On the basis of these two criteria, it is shown that crack growth can occur if 1) the applied stress increases, leading to \({K}_{I}\) to approach \({K}_{IC}\), 2) the crack reaches a critical length, leading to \({K}_{I}\) and \({K}_{IC}\) to approach each other, or 3) the surface energy decreases, leading to a reduction of \({K}_{IC}\) and hence a lower applied stress required to satisfy \({K}_{I}={K}_{IC}\). The latter can be achieved by changing the chemical environment and the concomitant adsorption of chemical species to the crack tip (Orowan 1944), as discussed below.
4.3.2 Crack Propagation in the Presence of Aqueous Fluids
In the presence of fluid, less work is required to produce new fracture surfaces (Parks 1984; Lawn 1993). This is due to the adsorption of chemically active molecules, such as water, to the Si–O bonds at the tip, which is made easier by stress concentrations at the strained tip of a stressed crack (Michalske and Freiman 1982; Michalske and Bunker 1984; Lindsay et al. 1994). After adsorption, the water molecule hydrolyses the Si–O bond, resulting in two silanol (> SiOH) surface groups, given as
where < or > indicate surface-bound species (Charles 1958; Dove 1995). Similarly, hydroxyl groups can corrode the Si–O bond according to
The newly formed silanol surface groups are weakly bonded by hydrogen bonds, requiring less energy to break than the original Si–O bond, aiding crack propagation at stress intensity levels below \({K}_{IC}\) and giving rise to subcritical crack growth, termed stress corrosion cracking (Atkinson and Meredith 1981; Michalske and Freiman 1982). The rate of crack growth is determined by the rate of the crack tip reaction, which in turn depends on the kinetics of the interfacial reaction mechanism or on the kinetics of transport of the reactive species to the moving crack tip (Atkinson 1984).
Note that the fracturing of quartz leads to the formation of free Si–O bonds extending from the solid surface (Parks 1984). The adsorption of chemical species to these free bonds on the quartz surface lowers the surface energy, by up to an order of magnitude in the presence of water, compared to vacuum conditions (Parks 1984). In solution, adsorption causes the quartz surface to be covered by positively (SiOH2+) and negatively (SiO−) charged surface complexes. Depending on solution pH, positive groups or negative groups dominate, or they are balanced, resulting in a positively, negatively or neutrally charged quartz surface, respectively (Parks 1984; Dove and Elston 1992; Du et al. 1994). At the point of zero charge, the surface energy is maximum (Parks 1984). For quartz, the pH of zero surface charge (pHPZC) is approximately 2–3 at room temperature (Parks 1965; Kosmulski 2018), though it is expected to be relatively temperature insensitive (Sverjensky and Sahai 1998). At solution pH > 4, the quartz surface charge is slightly negative, inducing a small attraction force on corrosive species. At higher solution pH (> 9), positive surface groups are rare (Du et al. 1994) and a pervasive negative surface charged is developed.
As surface energy and interfacial reaction rate are affected by the surface charge (Parks 1984), crack growth velocities in glass and quartz display a strong dependency on solution pH. Crack growth velocities increase with increasing solution pH above pH 4 and remain roughly constant below pH 2 (Wiederhorn and Johnson 1973; Atkinson and Meredith 1981; Dunning et al. 1984; Dove 1995). By contrast, in strong alkaline solutions crack growth is promoted by the abundance of hydroxyl groups, which are strongly corrosive compared to water (Casey et al. 1990; Dove 1995; see also Eq. 2). The presence, and adsorption, of other cations and anions may further impact the surface energy, as well as the crack propagation potential (Li and De Bruyn 1966; Parks 1984).
For crack growth below \({K}_{IC}\) in a surface-reactive environment, i.e., for subcritical crack growth by stress corrosion cracking, three distinct regimes are classically recognised (Fig. 10a; Atkinson 1984; Freiman 1984). In region 1, crack growth velocities are relatively slow and strongly depend on \({K}_{I}\) and the chemical activity (or partial pressure) of the pore fluid. Here, crack growth rates are controlled by an interfacial reaction rate at the crack tip. In region 2, the intermediate crack growth velocities are independent of \({K}_{I}\) and rate-limited by the transport of the corrosive species to the moving crack tip. In region 3, crack growth occurs at high velocities, reaching those of critical crack growth. Crack growth in this region is less well understood, but typically inferred to be dependent on some combination of chemically assisted and mechanical bond rupture. As a crack grows subcritically, its length and, consequently, \({K}_{I}\) at the crack tip increase. If the applied stress is fixed, this means that a crack initially characterised by region 1 behaviour can grow and propagate at increasingly higher velocities, thereby transitioning to region 2 and, subsequently, to region 3, to ultimately grow critically at \({K}_{I}={K}_{IC}\), resulting in grain failure (Fig. 10a).

a Schematic diagram for subcritical and critical crack growth in vacuum and chemically enhanced conditions, relating the crack growth velocity to stress intensity factor (\({K}_{I}\)) normalised to the critical stress intensity factor (\({K}_{IC}\)). \({K}_{0,i}\) denotes the threshold below which crack growth is presumed to cease in chemically enhanced and vacuum conditions. Region 1, 2 and 3 indicate reaction controlled, transport controlled and intrinsic bond rupture controlled crack growth, respectively. Points A, B and C indicate pathways where crack growth rates in a vacuum increase due to introduction of a corrosive fluid. b Crack growth frequency distribution outlining the areas of cracks growing critically, subcritically in vacuum conditions and subcritically in chemically enhanced conditions, and where crack growth is presumed to cease
In addition to these regions, there is a stress intensity threshold (\({K}_{0, chem}\), Fig. 10a) below which crack growth is presumed to cease. This threshold depends on chemical activity of the pore fluid and is found to strongly decrease with relative humidity for glass (Wiederhorn 1967; Kocer and Collins 2001). Considering the other similarities in crack growth in glass and quartz, a similar reduction in the crack growth threshold of quartz is expected.
In a vacuum, subcritical crack growth can still occur by intrinsic bond rupture processes (Wiederhorn et al. 1974), involving the growth of lattice trapped cracks through the motion of atomic kinks along the crack front (Thomson et al. 1971; Hsieh and Thomson 1973; Lawn 1975). When such a crack becomes saturated by a chemically active fluid, the crack growth process switches to stress corrosion cracking, producing an acceleration of crack growth rates as depicted in Fig. 10a, where crack growth velocities increase from point A to point B. Similarly, for cracks under vacuum conditions that are characterised by a stress intensity factor below \({K}_{0, vac}\) and therefore not propagating, fluid introduction may activate crack growth as illustrated by point C in Fig. 10a.
4.4 Effect of Chemical Environment on Deformation during Cyclic Compaction
4.4.1 Elastic Strain Evolution during Stress-Cycling
The current stress-cycling experiments allowed for a clear differentiation between total, permanent, and elastic strain (Fig. 5). From Fig. 5c, d, it is explicit that the pore fluid environment does not significantly influence the elastic deformation of the sand samples. This is not surprising as the elastic behaviour of a porous media under drained conditions (as employed in the current experiments) is mainly controlled by the elastic modulus of the sample material (Wang 2000). Furthermore, it can be seen that the amount of elastic strain accumulated per stress-cycle does slightly decrease with each subsequent cycle. This is likely related to densification of the aggregate, creating a stiffer aggregate (cf. Fig. 7a and b) and increasing the contact area between grains, thereby reducing the intergranular stresses, and hence the amount of elastic distortion.
4.4.2 Effect of Chemical Environment on Permanent Deformation during Stress-Cycling
Based on the theory of crack growth in low-vacuum and chemically active environments (cf. Sect. 4.3), we infer that the permanent deformation in our stress-cycling experiment was controlled by subcritical and critical crack growth, resulting in apparent time-independent deformation on the time scale investigated. It is assumed that after Stage I pre-compaction all samples contained a relatively similar distribution of cracks with specific lengths, reaching a specific \({K}_{I}\) during loading in the first stress-cycle of Stage II (Fig. 10b). During loading, for a small portion of these cracks \({K}_{I}={K}_{IC}\) (Fig. 10b). These cracks are presumed to grow critically, resulting in grain failure and permanent deformation. In addition, a portion of the distribution is likely to grow subcritically during loading, whereby the number of cracks growing subcritically depends on \({K}_{0}\) (Fig. 10b). Depending on the crack velocity, these subcritical cracks will grow fast enough to reach the critical crack length leading to critical failure or the applied stress is reduced before these conditions can be obtained.
The threshold for subcritical crack growth depends on the chemical environment and the resulting surface energy near the crack tip. The surface energy and \({K}_{0}\) are maximum in low-vacuum (dry) conditions (Wiederhorn 1967; Wan et al. 1990; Kocer and Collins 2001) and decrease in fluid-saturated conditions (Fig. 10b). In fluid-saturated conditions, the surface energy is maximum at the pHPZC and decreases with increasing solution pH (Parks 1984). Consequently, \({K}_{0}\) decreases with increasing solution pH and the number of crack growing subcritically during loading increases. The probability of cracks reaching the critical failure length and leading to permanent strain is, therefore, increasing going from low-vacuum (dry) conditions, to saturation with acidic solutions to alkaline solutions (Fig. 10b).
The distribution of cracks is presumably not stable during deformation. Cracks grown to failure, i.e., cracks which have produced grain failure, are no longer part of the distribution and the redistribution of interparticle stresses after grain failure is likely to change the stress field at the crack tip. It is therefore plausible that with progressive deformation the crack distribution changes, such that a smaller portion of the grains fail critically and more grow subcritically (Fig. 10b). As subcritical crack growth rates are significantly slower, more time is required for subcritical cracks to result in grain failure than would be the case for critical cracks. Consequently, the contribution of crack growth to deformation lessens with prolonged stress-cycling.
4.4.2.1 Permanent Deformation in Environments Devoid of Water
The environments devoid of water, i.e., low-vacuum and n-decane, showed the smallest amount of permanent strain, 0.7–0.9 and 0.9–1.0%, and AEs, 27,000–32,000 and 28,000–29,000 counts, in the first cycle, and low crack densities of 1.52 and 1.74 mm−2, respectively. Based on the difference between the two environments in cumulative AE count, which is a measure for grain rearrangement plus grain breakage, and crack density, which is a measure for grain breakage, it is inferred that in the presence of n-decane grain rearrangement played a smaller role during permanent deformation and the amount of volumetric strain accompanying grain breakage is therefore most likely larger than in the low-vacuum (dry) environment. Though n-decane is not an aqueous fluid, sample flooding did lead to an increase in compaction, which could perhaps be explained by a direct surface energy effect in the presence of fluid. Alternatively, trace amounts of water may have been present in the n-decane fluid, enhancing grain breakage, as already small amounts of water can enhance crack growth (Wiederhorn 1967). Furthermore, if trace amounts of water were present, then hydrolysis of the crack tip would lower the energy required for crack propagation and hence the energy released, which may be below the trigger threshold of our AE system, leading to an underestimation of the number of grain breakage events.
4.4.2.2 Permanent Deformation in Aqueous Solutions
A clear effect of aqueous solutions on permanent strain was observed in the first stress-cycle (Fig. 5e, f). Addition of distilled water resulted in 1.9% permanent strain, while less permanent strain (at least 1.0%) was measured in samples saturated with acidic solutions and more, up to 2.7% permanent strain, in alkaline solutions. Other studies on loose, non-pre-compacted, aggregates have shown that the chemical environment already impacts total strain near-instantaneously during loading, which then persists during creep (Hangx et al. 2010; Brzesowsky et al. 2014a). Similar behaviour is seen in pre-compacted samples, where fluid injection under applied stress immediately leads to an increase in strain (Brzesowsky et al. 2014a; Schimmel et al. 2019a), with the amount of strain being dependent on fluid composition. In our current study, the effect of fluid pH on total strain was partly overprinted by the slight decrease in elastic strain with increasing cycle number (cf. Sect. 4.2 and Fig. 5).
The trend in AE data with solution pH roughly correlates with the trend observed for permanent strain, with the exception of the pH 14 experiments (Fig. 5e, f and g, h). In the high alkaline solution, a lower than expected AE count was observed, while the permanent strain was markedly higher than in less alkaline environments. This could suggest that compaction mechanisms were active that do not produce AEs, like dissolution and/or pressure solution, as well as lubrication effects of the fluid leading to easier grain re-arrangement. Since none of the pore fluids were saturated with respect to quartz at the start of the experiment (cf. Sect. 2.1), the enhanced solubility and dissolution rate of quartz at higher pH (Brady and Walther 1990; Dove and Elston 1992) means that for the higher pH fluids (pH 9 and pH 14) more quartz could have dissolved during the experiment. This is particularly likely in the pH 14 experiments, as these samples were slightly cohesive after deformation. However, the strong reduction in grain size and high crack density (Fig. 8) do indicate that grain breakage was prevalent in these samples. The relatively low AE count may therefore be an effect undersampling of the AE events, caused by the reduced release of energy when cracks propagate in strong corrosive environments. In addition, dissolution of the surface of the quartz grains may have led to the formation of a gel-like layer (Dove and Crerar 1990), which could have promoted grain slip and hence rearrangement.
4.5 Implications
The current observations may have implications for porous subsurface reservoirs at 2–4 km depth with short-term or cyclic fluid injection, assuming that the same mechanism, i.e., microcracking, is operative. Though the stress changes applied in the current experiments are larger than the stress changes typically observed during reservoir injection-production operations, our results provide a worst-case scenario impression of prolonged, seasonal or temporary subsurface use. It should be noted that similar effects of chemical environment on time-dependent compaction (creep), representative for longer-term behaviour in the absence of pore pressure cycling, were observed (Schimmel et al. 2019a).
Geothermal energy production requires injection of cold fluid in chemical disequilibrium with the reservoir rock. Additives, such as scaling-inhibitors (like amino trimethylene phosphonic acid, or AMP), can be acidic (Knepper 2003; Finster et al. 2015) and may enhance any disequilibria effects. Our results suggest that an acidic environment will most likely not enhance any microcracking mechanisms in the quartz grains within a sandstone reservoir. However, it should be noted that we did not investigate the effect of acidic fluid injection into reservoirs containing other minerals, such as feldspars, clays or carbonate minerals. Depending on the load-bearing framework, dissolution of carbonate cements by acidification of the pore fluid may or may not weaken the rock (see e.g.,Le Guen et al. 2007; Hangx et al. 2013; Zhou et al. 2016). This in turn can affect the transport behaviour of the reservoir, impacting geothermal fluid production and injection. Therefore further research is needed on this.
Seasonal energy storage in depleted hydrocarbon reservoirs involves the injection of synthetic fuel, natural gas, hydrogen or compressed air, i.e., fluids that are predominantly devoid of water. Injection will induce changes in the stress and temperature field, and affect the chemical environment. Connate formation water may dissolve into the injected fluid, (locally) drying the reservoir. Our results show that continuous stress-cycling under low-vacuum (dry, i.e., comparable to hydrogen, gas or air storage) or inert (n-decane, i.e., comparable to synthetic fuel storage) conditions will minimise crack growth in the quartz grains within a sandstone. Furthermore, the inhibiting effect of an acidic environment on microcracking mechanisms in quartz grains suggest that temporary storage of CO2 for the generation of synthetic fuels will only have a limited effect on the mechanical behaviour of the reservoir (Jiang et al. 2010). As the environment may progressively become devoid of water, any dissolution/precipitation processes will also be limited.
Our results imply that on the short term, injection of alkaline wastewater in quartz-rich reservoirs could potentially lead to enhanced microcracking and/or stress-enhanced dissolution of the quartz grains, which may impact the mechanical behaviour of the reservoir. The amount of reservoir deformation that could be associated with the injection of alkaline fluid will depend on the mineralogical composition of the reservoir as well, and dissolution/precipitation processes should at least come to a halt when a new chemical equilibrium is reached. Though experiments have shown that on the long term, alkaline fluids will continue to drive stress corrosion cracking in quartz grains (Hangx et al. 2010; Schimmel et al. 2019a), more research would be needed to investigate the longer term behaviour of the reservoir in this context.
5 Conclusions
Uniaxial (oedometer) stress-cycling experiments were conducted to investigate the effect of pore fluid chemistry on time-independent compaction behaviour of quartz sand. The tests, performed at 80 °C and a maximum applied stress of 35 MPa, i.e., realistic reservoir conditions, involved a low-vacuum (dry) pre-compaction stage and a stress-cycling stage at either dry or fluid-saturated conditions. The employed fluids included an inert fluid (n-decane), distilled water and a selection of HCl and NaOH solutions covering a pH range of 1–14. Pre-compaction revealed that the amount of pre-compaction strain and AE depends on the sample starting porosity, where relatively high porosity samples produce more strain and AEs. Subsequent stress-cycling indicated the following effects:
-
1.
The majority of the strain accumulated in the first few stress-cycles and with each subsequent cycle the amount of total, permanent and elastic strain decreased. While permanent strain rapidly decreased from 0.7–2.7% towards an approximately constant value of 0.2–0.4% after ten stress-cycles, elastic stain only slightly lessened with prolonged stress-cycling. AE count also decreased with each stress-cycle from 27,000–65,000 in the first cycle to approximately 4,000 in the tenth stress-cycle.
-
2.
Introduction of a pore fluid affected the development of permanent and, hence, total strain, but had no influence on the elastic strain. Inert fluid (n-decane) slightly promoted compaction compared to low-vacuum (dry) conditions, while a more pronounced effect was observed for the addition of distilled water and aqueous solutions. AE count also increased in the presence of water-bearing fluids, except for a strongly alkaline solution (pH 14).
-
3.
The development of permanent strain roughly correlated with solution pH. Alkaline solutions promoted compaction and acidic solutions inhibited compaction compared to distilled water. This was underpinned by the grain size and crack density analyses that yielded smaller grain sizes and higher crack densities for alkaline solutions compared to acidic solutions.
-
4.
The sample stiffness was consistently lower for fluid-saturated samples compared to dry ones. Within one loading step, sample stiffness was initially low and increased to a peak value around 15 MPa sample stress, and either reduced again as observed in the first stress-cycle, or remained stable until approximately 26 MPa sample stress as seen in higher stress-cycles.
-
5.
It was inferred that elastic compaction was controlled by elastic deformation of grains and grain contacts. As the sample compacted, the coordination number of each grain increased, reducing the intergranular stresses and the driving force for elastic deformation. Permanent deformation was controlled by grain rearrangement during the initial part of loading, as evidenced by stiffening of the sample. Unlocking of the aggregate was achieved through grain breakage, which was aided by Si–O bond hydrolysis and pH-dependent fluid–solid surface interactions. With prolonged stress-cycling the effect of chemical environment decreased, suggesting that crack growth velocities were too slow to result in grain failure and, hence, permanent compaction.
-
6.
The results suggested that fluid injection into porous subsurface reservoirs could evoke an instantaneous chemical response, which could have favourable or negative consequences for reservoir performance, depending on the type of fluid injected and the host rock mineralogy. Since our study solely focused on the effect of fluid pH on the compaction of sand aggregates, more research is needed to study the impact for sandstones, both clean and impure.
Availability of Data and Material
The data for this paper are available as Schimmel et al. (2020) Stress-cycling data uniaxial compaction of quartz sand in various chemical environments. Data publication platform of Utrecht University. https://doi.org/10.24416/UU01-VM3Z6I.
References
Andričević R, Gotovac H, Lončar M, Srzić V (2009) Risk assessment from the oil waste disposal in deep wells. WIT Trans Built Environ 108:185–193. https://doi.org/10.2495/SAFE090181
Atkinson BK (1984) Subcritical crack growth in geological materials. J Geophys Res Solid Earth 89:4077–4114. https://doi.org/10.1029/JB089iB06p04077
Atkinson BK, Meredith PG (1981) Stress corrosion cracking of quartz: a note on the influence of chemical environment. Tectonophysics 77:T1–T11. https://doi.org/10.1016/0040-1951(81)90157-8
Bernabé Y, Fryer DT, Hayes JA (1992) The effect of cement on the strength of granular rocks. Geophys Res Lett 19:1511–1514. https://doi.org/10.1029/92GL01288
Brady PV, Walther JV (1990) Kinetics of quartz dissolution at low temperatures. Chem Geol 82:253–264. https://doi.org/10.1016/0009-2541(90)90084-K
Brantut N, Baud P, Heap MJ, Meredith PG (2012) Micromechanics of brittle creep in rocks. J Geophys Res Solid Earth. https://doi.org/10.1029/2012JB009299
Brzesowsky RH, Spiers CJ, Peach CJ, Hangx SJT (2011) Failure behavior of single sand grains: Theory versus experiment. J Geophys Res 116:B06205. https://doi.org/10.1029/2010JB008120
Brzesowsky RH, Hangx SJT, Brantut N, Spiers CJ (2014) Compaction creep of sands due to time-dependent grain failure: Effects of chemical environment, applied stress, and grain size. J Geophys Res Solid Earth 119:7521–7541. https://doi.org/10.1002/2014JB011277
Brzesowsky RH, Spiers CJ, Peach CJ, Hangx SJT (2014) Time-independent compaction behavior of quartz sands. J Geophys Res Solid Earth 119:936–956. https://doi.org/10.1002/2013JB010444
Budt M, Wolf D, Span R, Yan J (2016) A review on compressed air energy storage: Basic principles, past milestones and recent developments. Appl Energy 170:250–268. https://doi.org/10.1016/j.apenergy.2016.02.108
Casey WH, Lasaga AC, Gibbs GV (1990) Mechanisms of silica dissolution as inferred from the kinetic isotope effect. Geochim Cosmochim Acta 54:3369–3378. https://doi.org/10.1016/0016-7037(90)90291-R
Charles RJ (1958) Static Fatigue of Glass. I J Appl Phys 29:1549–1553. https://doi.org/10.1063/1.1722991
Chong S-H, Santamarina JC (2016) Sands subjected to repetitive vertical loading under zero lateral strain: accumulation models, terminal densities, and settlement. Can Geotech J 53:2039–2046. https://doi.org/10.1139/cgj-2016-0032
Chuhan FA, Kjeldstad A, Bjørlykke K, Høeg K (2002) Porosity loss in sand by grain crushing—experimental evidence and relevance to reservoir quality. Mar Pet Geol 19:39–53. https://doi.org/10.1016/S0264-8172(01)00049-6
Chuhan FA, Kjeldstad A, Bjørlykke K, Høeg K (2003) Experimental compression of loose sands: relevance to porosity reduction during burial in sedimentary basins. Can Geotech J 40:995–1011. https://doi.org/10.1139/t03-050
DeCastro M, Salvador S, Gómez-Gesteira M et al (2019) Europe, China and the United States: Three different approaches to the development of offshore wind energy. Renew Sustain Energy Rev 109:55–70. https://doi.org/10.1016/j.rser.2019.04.025
DeJong JT, Christoph GG (2009) Influence of particle properties and initial specimen state on one-dimensional compression and hydraulic conductivity. J Geotech Geoenvironmental Eng 135:449–454. https://doi.org/10.1061/(ASCE)1090-0241(2009)135:3(449)
Dewers TA, Hajash A (1995) Rate laws for water-assisted compaction and stress-induced water-rock interaction in sandstones. J Geophys Res 100:13093–13112. https://doi.org/10.1029/95JB00912
Doornhof D, Kristiansen TG, Nagel NB et al (2006) Compaction and subsidence. Oilf Rev 18:50–68
Dove PM (1995) Geochemical controls on the kinetics of quartz fracture at subcritical tensile stresses. J Geophys Res Solid Earth 100:22349–22359. https://doi.org/10.1029/95JB02155
Dove PM (1999) The dissolution kinetics of quartz in aqueous mixed cation solutions. Geochim Cosmochim Acta 63:3715–3727. https://doi.org/10.1016/S0016-7037(99)00218-5
Dove PM, Crerar DA (1990) Kinetics of quartz dissolution in electrolyte solutions using a hydrothermal mixed flow reactor. Geochim Cosmochim Acta 54:955–969. https://doi.org/10.1016/0016-7037(90)90431-J
Dove PM, Elston SF (1992) Dissolution kinetics of quartz in sodium chloride solutions: Analysis of existing data and a rate model for 25°C. Geochim Cosmochim Acta 56:4147–4156. https://doi.org/10.1016/0016-7037(92)90257-J
Du Q, Freysz E, Shen YR (1994) Vibrational spectra of water molecules at quartz/water interfaces. Phys Rev Lett 72:238–241. https://doi.org/10.1103/PhysRevLett.72.238
Dunning JD, Petrovski D, Schuyler J, Owens A (1984) The effects of aqueous chemical environments on crack propagation in quartz. J Geophys Res Solid Earth 89:4115–4123. https://doi.org/10.1029/JB089iB06p04115
Einav I (2007) Breakage mechanics-Part I: Theory. J Mech Phys Solids 55:1274–1297. https://doi.org/10.1016/j.jmps.2006.11.003
Fang H, Ciatto A, Brock F (2016) U.S. Natural Gas Storage Capacity and Utilization Outlook
Finster M, Clark C, Schroeder J, Martino L (2015) Geothermal produced fluids: Characteristics, treatment technologies, and management options. Renew Sustain Energy Rev 50:952–966. https://doi.org/10.1016/j.rser.2015.05.059
Freiman SW (1984) Effect of chemical enviroments on slow crack growth in glasses and ceramics. J Geophys Res 89:4072–4076
Gibson-Poole CM, Edwards S, Langford RP, Vakarelov B (2008) Review of geological storage opportunities for carbon capture and storage (CCS) in Victoria. PESA East Australas Basins Symp III:455–473
Gratier JP, Guiguet R, Renard F et al (2009) A pressure solution creep law for quartz from indentation experiments. J Geophys Res 114:B03403. https://doi.org/10.1029/2008JB005652
Graves C, Ebbesen SD, Mogensen M, Lackner KS (2011) Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew Sustain Energy Rev 15:1–23. https://doi.org/10.1016/j.rser.2010.07.014
Griffith AA (1920) The phenomena of rupture and flow in solids. Philos Trans R Soc London, Ser A, Contain Pap a Math or Phys Character 221:163–198
Hagerty MM, Hite DR, Ullrich CR, Hagerty DJ (1993) One-dimensional high-pressure compression of granular media. J Geotech Eng 119:1–18
Hajash A, Carpenter TD, Dewers TA (1998) Dissolution and time-dependent compaction of albite sand: experiments at 100°C and 160°C in pH-buffered organic acids and distilled water. Tectonophysics 295:93–115. https://doi.org/10.1016/S0040-1951(98)00117-6
Hangx SJT, Spiers CJ, Peach CJ (2010) Creep of simulated reservoir sands and coupled chemical-mechanical effects of CO2 injection. J Geophys Res Solid Earth 115:B09205. https://doi.org/10.1029/2009JB006939
Hangx SJT, Van Der Linden A, Marcelis F, Bauer A (2013) The effect of CO2 on the mechanical properties of the Captain Sandstone: Geological storage of CO2 at the Goldeneye field (UK). Int J Greenh Gas Control 19:609–619. https://doi.org/10.1016/j.ijggc.2012.12.016
Hauer C, Wagner B, Aigner J et al (2018) State of the art, shortcomings and future challenges for a sustainable sediment management in hydropower: a review. Renew Sustain Energy Rev 98:40–55. https://doi.org/10.1016/j.rser.2018.08.031
Heap MJ, Faulkner DR, Meredith PG, Vinciguerra S (2010) Elastic moduli evolution and accompanying stress changes with increasing crack damage: implications for stress changes around fault zones and volcanoes during deformation. Geophys J Int 183:225–236. https://doi.org/10.1111/j.1365-246X.2010.04726.x
Heinemann N, Wilkinson M, Pickup GE et al (2012) CO2 storage in the offshore UK Bunter Sandstone Formation. Int J Greenh Gas Control 6:210–219. https://doi.org/10.1016/j.ijggc.2011.11.002
Hertz H (1882) Ueber die Berührung fester elastischer Körper. J für die reine und Angew Math. https://doi.org/10.1515/crll.1882.92.156
Hsieh C, Thomson R (1973) Lattice theory of fracture and crack creep. J Appl Phys 44:2051–2063. https://doi.org/10.1063/1.1662512
IPCC (2005) IPCC special report on carbon dioxide capture and storage. Cambridge University Press, Cambridge
Irwin GR (1958) Fracture. In: S. F (ed) Elasticity and Plasticity/Elastizität und Plastizität. Handbuch der Physik/Encyclopedia of Physics. Springer, Berlin, Heidelberg
Jaeger JC (1967) Failure of rocks under tensile conditions. Int J Rock Mech Min Sci 4:219–227. https://doi.org/10.1016/0148-9062(67)90046-0
Jiang Z, Xiao T, Kuznetsov VL, Edwards PP (2010) Turning carbon dioxide into fuel. Philos Trans R Soc A Math Phys Eng Sci 368:3343–3364. https://doi.org/10.1098/rsta.2010.0119
Juez-Larré J, Remmelts G, Breunese JN et al (2016) Using underground gas storage to replace the swing capacity of the giant natural gas field of Groningen in the Netherlands. A reservoir performance feasibility study. J Pet Sci Eng 145:34–53. https://doi.org/10.1016/j.petrol.2016.03.010
Kabir E, Kumar P, Kumar S et al (2018) Solar energy: Potential and future prospects. Renew Sustain Energy Rev 82:894–900. https://doi.org/10.1016/j.rser.2017.09.094
Keranen KM, Weingarten M, Abers GA et al (2014) Sharp increase in central Oklahoma seismicity since 2008 induced by massive wastewater injection. Science (80-) 345:448–451. https://doi.org/10.1126/science.1255802
Knepper T (2003) Synthetic chelating agents and compounds exhibiting complexing properties in the aquatic environment. Trends Anal Chem 22:708–724. https://doi.org/10.1016/S0165-9936(03)01008-2
Kocer C, Collins RE (2001) Measurement of very slow crack growth in glass. J Am Ceram Soc 84:2585–2593. https://doi.org/10.1111/j.1151-2916.2001.tb01058.x
Kosmulski M (2018) The pH dependent surface charging and points of zero charge. VII. Update. Adv Colloid Interface Sci 251:115–138. https://doi.org/10.1016/j.cis.2017.10.005
Lade PV, Yamamuro JA, Bopp PA (1996) Significance of particle crushing in granular materials. J Geotech Geoenviron Eng 122:309–316. https://doi.org/10.1061/(ASCE)1090-0241(1997)123:9(889)
Lawn BR (1975) An atomistic model of kinetic crack growth in brittle solids. J Mater Sci 10:469–480. https://doi.org/10.1007/BF00543692
Lawn BR (1993) Fracture of brittle solids. Cambridge University Press, Cambridge
Le Guen Y, Renard F, Hellmann R et al (2007) Enhanced deformation of limestone and sandstone in the presence of high Pco2 fluids. J Geophys Res 112:B05421. https://doi.org/10.1029/2006JB004637
Lehner FK (1990) Thermodynamics of rock deformation by pressure solution. Deformation processes in minerals, ceramics and Rocks. Springer, Netherlands, Dordrecht, pp 296–333
Lei X, Ma S, Chen W et al (2013) A detailed view of the injection-induced seismicity in a natural gas reservoir in Zigong, southwestern Sichuan Basin, China. J Geophys Res Solid Earth 118:4296–4311. https://doi.org/10.1002/jgrb.50310
Li HC, De Bruyn PL (1966) Electrokinetic and adsorption studies on quartz. Surf Sci 5:203–220. https://doi.org/10.1016/0039-6028(66)90082-3
Limberger J, Boxem T, Pluymaekers M et al (2018) Geothermal energy in deep aquifers: A global assessment of the resource base for direct heat utilization. Renew Sustain Energy Rev 82:961–975. https://doi.org/10.1016/j.rser.2017.09.084
Lindsay CG, White GS, Freiman SW, Wong-Ng W (1994) Molecular orbital study of an environmentally enhanced crack growth process in silica. J Am Ceram Soc 77:2179–2187. https://doi.org/10.1111/j.1151-2916.1994.tb07115.x
Liu W, Ramirez A (2017) State of the art review of the environmental assessment and risks of underground geo-energy resources exploitation. Renew Sustain Energy Rev 76:628–644. https://doi.org/10.1016/j.rser.2017.03.087
Mahlia TMI, Saktisahdan TJ, Jannifar A et al (2014) A review of available methods and development on energy storage; technology update. Renew Sustain Energy Rev 33:532–545. https://doi.org/10.1016/j.rser.2014.01.068
Mao C, Yamada Y, Matsuoka T (2014) A preliminary assessment of geological CO2 storage in Cambodia. Int J Greenh Gas Control 30:19–33. https://doi.org/10.1016/j.ijggc.2014.08.016
McDowell GR, Bolton MD (1998) On the micromechanics of crushable aggregates. Géotechnique 48:667–679. https://doi.org/10.1680/geot.2000.50.3.315
Mesri G, Vardhanabhuti B (2009) Compression of granular materials. Can Geotech J 46:369–392. https://doi.org/10.1139/T08-123
Michalske TA, Bunker BC (1984) Slow fracture model based on strained silicate structures. J Appl Phys 56:2686–2693. https://doi.org/10.1063/1.333789
Michalske TA, Freiman SW (1982) A molecular interpretation of stress corrosion in silica. Nature 295:511–512. https://doi.org/10.1038/295511a0
Moeck IS (2014) Catalog of geothermal play types based on geologic controls. Renew Sustain Energy Rev 37:867–882. https://doi.org/10.1016/j.rser.2014.05.032
Monismith CL, Ogawa N, Freeme CR (1975) Permanent deformation characteristics of subgrade soils due to repeated loading. Transp Res Rec 537:17
Moriarty P, Honnery D (2016) Can renewable energy power the future? Energy Policy 93:3–7. https://doi.org/10.1016/j.enpol.2016.02.051
Nagel NB (2001) Compaction and subsidence issues within the petroleum industry: from Wilmington to Ekofisk and beyond. Phys Chem Earth, Part A Solid Earth Geod 26:3–14. https://doi.org/10.1016/S1464-1895(01)00015-1
Nakata Y, Hyodo M, Hyde AFL et al (2001) Microscopic particle crushing of sand subjected to high pressure one-dimensional compression. Soils Found 41:69–82. https://doi.org/10.3208/sandf.41.69
Nakata Y, Kato Y, Hyodo M et al (2001) One-dimensional compression behaviour of uniformly graded sand related to single particle crushing strength. Soils Found 41:39–51
Omidvar M, Iskander M, Bless S (2012) Stress-strain behavior of sand at high strain rates. Int J Impact Eng 49:192–213. https://doi.org/10.1016/j.ijimpeng.2012.03.004
Orowan E (1944) The fatigue of glass under stress. Nature 154:341–343. https://doi.org/10.1038/154341a0
Park J, Santamarina JC (2018) Sand response to a large number of loading cycles under zero-lateral-strain conditions: evolution of void ratio and small-strain stiffness. Géotechnique. https://doi.org/10.1680/jgeot.17.p.124
Parks GA (1965) The isoelectric points of solid oxides, solid hydroxides, and aqueous hydroxo complex systems. Chem Rev 65:177–198. https://doi.org/10.1021/cr60234a002
Parks GA (1984) Surface and interfacial free energies of quartz. J Geophys Res 89:3997–4008. https://doi.org/10.1029/JB089iB06p03997
Pluymakers AMH, Peach CJ, Spiers CJ (2014) Diagenetic compaction experiments on simulated anhydrite fault gouge under static conditions. J Geophys Res Solid Earth 119:4123–4148. https://doi.org/10.1002/2014JB011073
Potyondy DO, Cundall PA (2004) A bonded-particle model for rock. Int J Rock Mech Min Sci 41:1329–1364. https://doi.org/10.1016/j.ijrmms.2004.09.011
Rice JR (1978) Thermodynamics of the quasi-static growth of Griffith cracks. J Mech Phys Solids 26:61–78. https://doi.org/10.1016/0022-5096(78)90014-5
Rutter EH (1983) Pressure solution in nature, theory and experiment. J Geol Soc London 140:725–740. https://doi.org/10.1144/gsjgs.140.5.0725
Sawicki A, Swidzinski W (1995) Cyclic compaction of soils, grains and powders. Powder Technol 85:97–104. https://doi.org/10.1016/0032-5910(95)03013-Y
Schimmel MTW, Hangx SJT, Spiers CJ (2019) Impact of chemical environment on compaction creep of quartz sand and possible geomechanical applications. J Geophys Res Solid Earth 124:5584–5606. https://doi.org/10.1029/2019JB017464
Schimmel MTW, Liu W, Worrell E (2019) Facilitating sustainable geo-resources exploitation: a review of environmental and geological risks of fluid injection into hydrocarbon reservoirs. Earth-Sci Rev 194:455–471. https://doi.org/10.1016/j.earscirev.2019.03.006
Schutjens PMTM (1991) Experimental compaction of quartz sand at low effective stress and temperature conditions. J Geol Soc London 148:527–539. https://doi.org/10.1144/gsjgs.148.3.0527
Shortall R, Davidsdottir B, Axelsson G (2015) Geothermal energy for sustainable development: a review of sustainability impacts and assessment frameworks. Renew Sustain Energy Rev 44:391–406. https://doi.org/10.1016/j.rser.2014.12.020
Spiers CJ, De Meer S, Niemeijer AR, Zhang X (2004) Kinetics of rock deformation by pressure solution and the role of thin aqueous films. In: Nakashima S, Spiers CJ, Mercury L et al (eds) Physicochemistry of water in geological and biological systems. Universal Academy Press Inc, Tokyo, pp 129–158
Suckale J (2009) Induced seismicity in hydrocarbon fields. Adv Geophys 51:55–106. https://doi.org/10.1016/S0065-2687(09)05107-3
Sverjensky DA, Sahai N (1998) Theoretical prediction of single-site enthalpies of surface protonation for oxides and silicates in water. Geochim Cosmochim Acta 62:3703–3716. https://doi.org/10.1016/S0016-7037(98)00262-2
Takei M, Kusakabe O, Hayashi T (2001) Time-dependent behavior of crushable materials in one-dimensional compression tests. Soils Found 41:97–121. https://doi.org/10.3208/sandf.41.97
Tarkowski R (2019) Underground hydrogen storage: characteristics and prospects. Renew Sustain Energy Rev 105:86–94. https://doi.org/10.1016/j.rser.2019.01.051
Thomson R, Hsieh C, Rana V (1971) Lattice trapping of fracture cracks. J Appl Phys 42:3154–3160. https://doi.org/10.1063/1.1660699
Underschultz J, Boreham C, Dance T et al (2011) CO2 storage in a depleted gas field: an overview of the CO2CRC Otway Project and initial results. Int J Greenh Gas Control 5:922–932. https://doi.org/10.1016/j.ijggc.2011.02.009
Wan K, Lathabai S, Lawn BR (1990) Crack velocity functions and thresholds in brittle solids. J Eur Ceram Soc 6:259–268. https://doi.org/10.1016/0955-2219(90)90053-I
Wang HF (2000) Theory of linear poroelasticity with applications to geomechanics and hydrogeology. Princeton University Press, New Jersey
Wang Z, Wong RCK, Qiao L (2011) Investigation on relations between grain crushing amount and void ratio change of granular materials in one-dimensional compression and creep tests. J Rock Mech Geotech Eng 3:415–420. https://doi.org/10.3724/SP.J.1235.2011.00415
Wichtmann T, Niemunis A, Triantafyllidis T (2005) Strain accumulation in sand due to cyclic loading: drained triaxial tests. Soil Dyn Earthq Eng 25:967–979. https://doi.org/10.1016/j.soildyn.2005.02.022
Wiederhorn SM (1967) Influence of Water Vapor on Crack Propagation in Soda-Lime Glass. J Am Ceram Soc 50:407–414. https://doi.org/10.1111/j.1151-2916.1967.tb15145.x
Wiederhorn SM, Johnson H (1973) Effect of electrolyte pH on crack propagation in glass. J Am Ceram Soc 56:192–197. https://doi.org/10.1111/j.1151-2916.1973.tb12454.x
Wiederhorn SM, Johnson H, Diness AM, Heuer AH (1974) Fracture of glass in vacuum. J Am Ceram Soc 57:336–341. https://doi.org/10.1111/j.1151-2916.1974.tb10917.x
Yamamuro JA, Bopp PA, Lade PV (1996) One-dimensional compression of sands at high pressures. J Geotech Eng 122:147–154. https://doi.org/10.1061/(ASCE)0733-9410(1996)122:2(147)
Zhang J, Wong T-F, Davis DM (1990) Micromechanics of pressure-induced grain crushing in porous rocks. J Geophys Res 95:341–352. https://doi.org/10.1029/JB095iB01p00341
Zhou H, Hu D, Zhang F et al (2016) Laboratory investigations of the hydro-mechanical–chemical coupling behaviour of sandstone in CO2 storage in aquifers. Rock Mech Rock Eng 49:417–426. https://doi.org/10.1007/s00603-015-0752-8
Acknowledgements
This work is part of the research programme Transitioning to a More Sustainable Energy System with project number 022.004.023 which is financed by the Netherlands Organisation for Scientific Research (NWO). Declarations of interest: none. The technical staff of the HPT Laboratory is thanked for their assistance, especially Gert Kastelein, Eimert de Graaff, Thony van der Gon-Netscher and Floris van Oort. Leonard Bik is thanked for preparing the thin sections. We are thankful to Colin Peach and Tim Wolterbeek for assistance in the lab and constructive discussions. We also thank two anonymous reviewers for their feedback, which helped to improve the manuscript. The data for this paper are available as Schimmel et al. (2020) Stress-cycling data uniaxial compaction of quartz sand in various chemical environments. Data publication platform of Utrecht University. https://doi.org/10.24416/UU01-VM3Z6I.
Funding
This work is part of the research programme Transitioning to a More Sustainable Energy System with project number 022.004.023, which is financed by the Netherlands Organisation for Scientific Research (NWO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The work was performed at Experimental Rock Deformation, Department of Earth Sciences, Faculty of Geosciences, Utrecht University.
Rights and permissions
About this article

Cite this article
Schimmel, M.T.W., Hangx, S.J.T. & Spiers, C.J. Impact of Chemical Environment on Compaction Behaviour of Quartz Sands during Stress-Cycling. Rock Mech Rock Eng 54, 981–1003 (2021). https://doi.org/10.1007/s00603-020-02267-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00603-020-02267-0