
Abstract
Arbuscular mycorrhizal (AM) fungi form a root endosymbiosis with many agronomically important crop species. They enhance the ability of their host to obtain nutrients from the soil and increase the tolerance to biotic and abiotic stressors. However, AM fungal species can differ in the benefits they provide to their host plants. Here, we examined the putative molecular mechanisms involved in the regulation of the physiological response of Medicago truncatula to colonization by Rhizophagus irregularis or Glomus aggregatum, which have previously been characterized as high- and low-benefit AM fungal species, respectively. Colonization with R. irregularis led to greater growth and nutrient uptake than colonization with G. aggregatum. These benefits were linked to an elevated expression in the roots of strigolactone biosynthesis genes (NSP1, NSP2, CCD7, and MAX1a), mycorrhiza-induced phosphate (PT8), ammonium (AMT2;3), and nitrate (NPF4.12) transporters and the putative ammonium transporter NIP1;5. R. irregularis also stimulated the expression of photosynthesis-related genes in the shoot and the upregulation of the sugar transporters SWEET1.2, SWEET3.3, and SWEET 12 and the lipid biosynthesis gene RAM2 in the roots. In contrast, G. aggregatum induced the expression of biotic stress defense response genes in the shoots, and several genes associated with abiotic stress in the roots. This suggests that either the host perceives colonization by G. aggregatum as pathogen attack or that G. aggregatum can prime host defense responses. Our findings highlight molecular mechanisms that host plants may use to regulate their association with high- and low-benefit arbuscular mycorrhizal symbionts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrogen (N) and phosphorus (P) are the main nutrients that limit plant growth in natural and agricultural ecosystems (LeBauer and Treseder 2008; Vitousek et al. 2010). Conventional management practices typically address both N and P limitations through the application of chemical fertilizers, but fertilizer production, transportation, and application are both economically and environmentally expensive (Havlin et al. 2014). Implementing alternative sustainable methods to mitigate nutrient deficiencies in crops is essential to prevent continued environmental degradation and to meet the nutritional demands of a growing human population.
To overcome nutrient limitations, plants have evolved the capacity to form alliances with mutualistic, root-associated microbes (Martin et al. 2017). The most widespread mutualism is the arbuscular mycorrhizal (AM) symbiosis formed by 72% of all known plant species and a relatively small number of fungal species in the Glomeromycotina and Mucoromycotina (Spatafora et al. 2016; Brundrett and Tedersoo 2018). AM fungi provide the host plant with mineral nutrients, such as P and N, and improve the resistance of plants against abiotic and biotic stressors (Bücking and Kafle 2015; Kafle et al. 2019).
Under P deficiency, plants increase the production of strigolactones, a class of phytohormones that also serve as signaling molecules perceived by AM fungi (Yoneyama et al. 2007). Strigolactones are exuded into the rhizosphere through the ATP-binding cassette transporter PDR1 (Kretzschmar et al. 2012). AM fungal perception of strigolactones triggers spore germination, hyphal branching, and the production of short-chain chitin oligomers (Besserer et al. 2008; Genre et al. 2013). These, along with lipo-chitooligosaccharides (Maillet et al. 2011; Rush et al. 2020), comprise the so-called Myc factors that prime the host plant for AM root colonization by activating the “common symbiosis signaling pathway” (CSSP; reviewed in MacLean et al. 2017).
The key steps of the CSSP include (1) the perception of Myc factors at the plasma membrane of root epidermal cells by lysine-motif receptor-like kinases (e.g., NFP and CERK1; Feng et al. 2019) and the leucine-rich repeat receptor-like kinase DMI2/SYMRK (Stracke et al. 2002); (2) the activation of the secondary messenger mevalonate that likely activates multiple nuclear membrane-localized calcium channels (CASTOR, DMI1/POLLUX, and CNGC15; Venkateshwaran et al. 2015; Kim et al. 2019b); (3) oscillations in calcium ion concentration in the nucleoplasm that activate calcium- and calmodulin-dependent protein kinase (DMI3/CCaMK; Lévy et al. 2004; Singh and Parniske 2012); (4) phosphorylation of IPD3/CYCLOPS which, in concert with DELLA, induces the expression of RAM1 (Horváth et al. 2011; Pimprikar et al. 2016); and (5) activation of several other transcription factors required for the development and regulation of the AM symbiosis (Pimprikar and Gutjahr 2018).
Following root penetration, AM hyphae proliferate both intra- and intercellularly and form highly branched nutrient exchange structures called arbuscules in cortical cells (Park et al. 2015). Surrounding the arbuscule is a plant-derived periarbuscular membrane (PAM) that is enriched in AM-induced transporters that facilitate reciprocal nutrient exchange between the fungus and the plant (Garcia et al. 2016). In Medicago truncatula, three AMT2 family ammonium transporters (AMT2;3, AMT2;4, and AMT2;5; Breuillin-Sessoms et al. 2015) and the phosphate transporters PT4 (Javot et al. 2007) and PT8 (Breuillin-Sessoms et al. 2015) are involved in the uptake of N and P from the periarbuscular space between the fungal plasma membrane and the PAM. In exchange, host plants transfer between 4 and 20% of their photosynthetically derived carbon to their fungal partners, and these high carbon costs force host plants to strongly regulate the carbon flux to their fungal symbionts (Paul and Kucey 1981; Wright et al. 1998; Lendenmann et al. 2011). The host plant releases lipids and sugars into the mycorrhizal interface likely via the putative lipid transporters STR and STR2 (Zhang et al. 2010; Roth and Paszkowski 2017) and the bidirectional sugar transporter SWEET1.2 (formerly SWEET1b, An et al. 2019; Doidy et al. 2019).
AM fungal species differ in the benefits they provide to their host plant. As observed previously in several plant species, the cooperative AM fungus Rhizophagus irregularis 09 (Ri09) transfers more N and P to its host plant and contributes to higher mycorrhizal growth responses than the less-cooperative fungus Glomus aggregatum 165 (Ga165; Kiers et al. 2011; Fellbaum et al. 2012, 2014; Wang et al. 2016). Symbiosis with Ga165 is also more costly for the host plant (measured as carbon costs per P transferred) than that with Ri09, and M. truncatula preferentially allocates more carbon to Ri09 than to Ga165 (Kiers et al. 2011). Collectively, these studies suggest that carbon to nutrient exchange processes are driven by biological market dynamics, but the molecular mechanisms that regulate these processes are largely unknown. We hypothesized that the host plant regulates the expression of key genes in response to colonization by AM fungi that differ in the nutrient benefits that they provide for the host. To test this hypothesis, we examined the physiological and transcriptomic responses of the host plant M. truncatula to colonization by either high- (Ri09) or low-benefit (Ga165) AM fungal partners under N and P limitation.
Materials and methods
Experimental design
Custom-made, two-compartment pots (12 cm × 8 cm × 8 cm, L × W × H; Supplemental Fig. S1) were used to physically isolate the roots of Medicago truncatula in a root compartment (RC) from extraradical hyphae of AM fungi in a hyphal compartment (HC). The RC was separated from the HC by a 0.1-cm-thick plastic divider sealed on all sides by silicone (Aqueon, Franklin, WI, USA). A hole in the middle of the divider (~ 3.12 cm in diameter) was covered on both sides with a sheet of fine nylon mesh with 50-µm pores. Between these sheets was a coarse nylon mesh with 1000-µm pores to create an air gap and inhibit mass flow between the RC and the HC. This allowed fungal hyphae, but not roots, to crossover from the RC to the HC (Supplemental Fig. S1), thus enabling an independent evaluation of how well Ga165 and Ri09 individually capture N and P in the HC and deliver it to the roots of the host plant in the RC. Using these split-compartment pots, two experiments with a similar design but different harvest times were performed. As described in detail below, the plants in both independent experiments were inoculated with either Ri09 or Ga165, or mock-inoculated, and were harvested 3 or 7 weeks post-inoculation (WPI), respectively. The number of independent biological replicates (plants) at 3 or 7 WPI were three and five, respectively.
Biological materials and growth conditions
M. truncatula “Jemalong A17” seeds were acid scarified in 36 N H2SO4 (Fisher Scientific Inc., Waltham, MA, USA), surface sterilized with 8.25% sodium hypochlorite (Clorox® bleach, The Clorox Company, Oakland, CA, USA), rinsed with sterile Type 1 water, and imbibed at 4 °C overnight. The seeds were then transferred onto moist autoclaved filter paper in Petri dishes and kept in the dark for 3 days. Finally, the Petri dishes were placed in ambient conditions for 7 days. Fully germinated seedlings were transferred into the RC of the two-compartment pots. Both the RC and HC were filled with 250 ml of sterilized soil substrate containing 40% sand, 20% perlite, 20% vermiculite, and 20% soil by volume. The soil substrate had original concentrations of 4.81 mg kg−1 of Olsen’s extractable phosphate, 10 mg kg−1 of NH4+, 34.4 mg kg−1 of NO3−, and a pH of 8.26. These nutrient levels were sufficiently low to induce host demand for N and P and to stimulate AM colonization.
Fungal inocula for Rhizophagus irregularis isolate 09 (Ri09; collected from Southwest Spain by Mycovitro S.L. Biotechnología Ecológica, Granada, Spain) and Glomus aggregatum isolate 165 (Ga165; collected from the Long Term Mycorrhizal Research Site, University of Guelph, Canada) were produced using axenic root organ cultures of Ri T-DNA–transformed carrot (Daucus carota clone DCI) grown on minimal medium (St-Arnaud et al. 1996). After approximately 8 weeks of growth, spores were isolated by blending the medium in 10 mM citrate buffer (pH 6.0). Then, at transplanting, each seedling was inoculated with ~ 0.4 g mycorrhizal roots and ~ 350 (plants harvested at 3 WPI) or ~ 500 (plants harvested at 7 WPI) spores of either Ri09 or Ga165. The difference in spore counts at 3 and 7 WPI was due to a slight variation in the abundance of spores in the root organ cultures from which the spores were isolated. Non-mycorrhizal (NM) controls received a similar quantity of double autoclaved roots and spores. After transplanting and fungal inoculation, the plants were grown in a growth chamber (model TC30; Conviron, Winnipeg, MB, Canada) with a 25 °C day/20 °C night cycle, 30% relative humidity, and a photosynthetic photon flux of ~ 225 μmol m−2 s−1 with a 16-h photoperiod. Two weeks before the plants of the 7 WPI experiment were harvested, 3.072 mmol of 15NH4Cl (Sigma Aldrich, St. Louis, USA) and 0.384 mmol of KH2PO4 were added to the HC of both inoculated and mock-inoculated plants in a modified Ingestad solution (Ingestad 1960). This resulted in final pot concentrations of 4 mM and 0.5 mM for 15NH4Cl and KH2PO4, respectively. Throughout the experiment, the position of the plants within the growth chamber was randomized to avoid positional effects.
Biomass determination, mycorrhizal quantification, and analysis of nitrogen and phosphorus contents in plants
At plant harvest, shoots and roots were separated to determine their respective fresh weight. Subsamples of both tissues were flash frozen in liquid N2 and stored at − 80 °C for transcriptome analysis. An additional root subsample was collected and stored in 50% ethanol at 4 °C to assess AM colonization. The remaining plant tissue was dried at 70 °C for 48 h, and then shoot and root dry weights were recorded. To determine the level of AM colonization, the root samples were cleared with 10% KOH at 80 °C for 30 min, rinsed with tap water, and stained with 5% Sheaffer ink-vinegar (v:v) at 80 °C for 15 min (Vierheilig et al. 1998). Total AM colonization of entire root subsamples cut into ~ 1-cm segments was assessed using the gridline intersection method (McGonigle et al. 1990). On average, ~ 110 root-gridline intersections were analyzed at 3 WPI (due to the smaller root size) and ~ 200 root-gridline intersections at 7 WPI. A root-gridline intersection was considered “colonized” if intraradical hyphae were present in the root at the site of the gridline intersection. Arbuscule and vesicle density also were determined by counting the number of root-gridline intersections where one or more arbuscules or vesicles were present. We did not observe any fungal structures in the roots of NM control plants. For plants harvested at 7 WPI, subsamples of dried shoot and root tissues were pulverized using a tissue homogenizer (Precellys 24 Dual, Cayman Chemical Company, Ann Arbor, MI, USA) and digested with 2 N HCl for 2 h at 95 °C. The P concentration in the plant tissues was spectrophotometrically determined at 436 nm after adding ammonium molybdate vanadate solution (Fisher Scientific, Pittsburgh, USA). Measurements of nitrogen-15 in plant tissues were performed by quantitative NMR spectroscopy as described previously (Kafle et al. 2018).
Fluorescent staining and confocal analysis of arbuscules
A selection of representative ink-stained roots from each biological replicate that contained arbuscules were subsequently stained for confocal imaging of hyphal structures as described in Cope et al. (2019). Briefly, they were incubated overnight at 4 °C in 1X PBS containing 2 µg ml−1 Alexa Fluor 488™ conjugated to wheat germ agglutinin (WGA; Thermo Fischer Scientific, Waltham, MA, USA). The next day, stained roots were rinsed with 1X PBS, mounted on a glass slide, and immediately observed on an Olympus FV1200 confocal laser scanning microscope (Olympus, Shinjuku, Tokyo, Japan) with an Olympus UPlanApo 40 × /0.85 objective. The emission spectra of Alexa Fluor 488™ WGA (500 to 540 nm) were detected after excitation with a 488-nm laser. Other settings used in the Fluoview v4.2c acquisition software (e.g., laser intensity, gain, offset, magnification, airy units) were similar for all sample observations. Using Fiji (Schindelin et al. 2012), we measured the width of 132 and 80 arbuscules from 11 Ga165-colonized root segments and 16 Ri09-colonized root segments, respectively, at the 3 WPI time point and 488 and 508 arbuscules from 22 Ga165-colonized root segments and 21 Ri09-colonized root segments, respectively, at the 7 WPI time point (the colonized root segments were collected from a minimum of 3 biological replicates per treatment).
RNA isolation and sequencing
Root and shoot tissues from the 7 WPI experiment were individually homogenized in liquid N2, and total RNA was extracted using the PureLink™ RNA Mini Kit (Thermo Fisher Scientific). Extracted RNAs were treated with TURBO™ DNase (Thermo Fisher Scientific) and quantified using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific). RNA quality was assessed using 2100 BioAnalyzer technology (Agilent Technologies). From each colonization regime, samples from four plants (n = 4) with both the highest yield and quality were used for further analysis. Approximately 100 ng of total RNA was used to construct poly(A) selection libraries using the Illumina TruSeq RNA Sample Preparation kit, and 150-nucleotide single-end reads were sequenced using an Illumina NextSeq500 with high output flow cells (Genomics Sequencing Facility, South Dakota State University).
The RNA sequencing data were uploaded to Discovery Environment (https://de.cyverse.org/de) and read quality of each fastq file was evaluated using FastQC 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). One Ga165 and one NM root sample and one Ri09 shoot sample were discarded because of poor quality. The remaining root (n = 3, Ga165 and NM; n = 4, Ri09) and shoot (n = 4, Ga165 and NM; n = 3, Ri09) samples were aligned to the reference genome of M. truncatula “Jemalong A17” (JCVI Mt4.0; Tang et al. 2014) using HISAT2.1 (Kim et al. 2019a). Aligned reads were then assembled and merged using StringTie-1.3.3 (Pertea et al. 2015) and the M. truncatula JCVI 4.0v2 annotation file (Tang et al. 2014). Finally, Cuffdiff2.1.1 (Trapnell et al. 2013) was used to quantify transcript abundance. The output files from CuffDiff2.1.1 were visualized in R v3.6.2 (The R Foundation 2020) using the CummeRbund package (Goff et al. 2012). The differential gene expression output files from CuffDiff2.1.1 included comparisons not only between Ri09 or Ga165 inoculated plants versus mock-inoculated plants, but also Ri09 versus Ga165. For further analysis, the CuffDiff2.1.1 output files were imported into Microsoft Excel (Microsoft 365®) and sorted to identify significantly differentially expressed genes (DEGs) among the three treatment comparisons with a log2 fold-change > 2 and a false discovery rate-adjusted q-value < 0.05. All DEGs outside this threshold were not considered for further analysis. Venn diagrams of DEGs were generated using the Bioinformatics & Evolutionary Genomics Web tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) and heatmaps of FPKM (fragments per kilobase of transcript per million mapped reads) values from all DEGs across inoculation regimes were generated using Cluster 3.0 and Java TreeView (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). Gene ontology (GO) enrichment analysis was conducted with the web-based tool PlantTFDB 4.0 (http://plantregmap.gao-lab.org/go.php).
Statistical analysis
For the physiological response variables at 7 WPI, unless mentioned otherwise, the mean and standard error of the mean were determined from five independent biological replicates for each colonization regime, including noncolonized and either Ga165- or Ri09-colonized plants. Both the Shapiro–Wilk normality and Levene’s homogeneity of variances tests were performed. Data sets that did not pass (p ≤ 0.05) were either log, square root, or square transformed to meet analysis assumptions. Then, one-way ANOVA was used to identify statistically significant differences (p ≤ 0.05) among all three colonization regimes for dry weight, P concentration, P content, and nitrogen-15 enrichment. The least significant difference (LSD) test was subsequently used for pairwise multiple comparisons to identify significant differences (p ≤ 0.05) among treatments. These statistical analyses were conducted using Statistix 9 (analytical software, Tallahassee, FL, USA). For AM colonization and arbuscule size, statistically significant differences (p ≤ 0.05) between the Ga165 and Ri09 mycorrhizal treatments were determined using a Welch’s two-sample t test in R v4.0.2 (The R Foundation 2020). Only results that are significant at the p ≤ 0.05 level are discussed. All F statistics and p values are shown in Supplemental Table S1.
Results
Colonization by Rhizophagus irregularis 09 leads to greater plant biomass and phosphorus and nitrogen uptake than that by Glomus aggregatum 165
We used a dual-compartment pot system to compare the overall benefit that Ga165 and Ri09 provided to M. truncatula (Supplemental Fig. S1). In plants grown for 3 WPI, root length colonization with arbuscules and vesicles was significantly lower in Ga165 than in Ri09-colonized roots (Supplemental Fig. S2a). There are indications that colonization with intraradical hyphae was also lower, but the difference was only near significance (p = 0.0873). In contrast, at 7 WPI, percent root length colonization by intraradical hyphae and arbuscules reached similar levels in roots colonized by either fungus (Supplemental Fig. S2b). Interestingly, the percentage of roots containing vesicles was significantly higher in Ri09-colonized roots at 3 WPI, but higher in Ga165-colonized roots at 7 WPI (Supplemental Fig. S2). Arbuscule size was significantly higher in Ri09-colonized roots at 3 WPI, but comparable between the AM fungal species at 7 WPI (Fig. 1).

Arbuscule size in Medicago truncatula roots inoculated with arbuscular mycorrhizal fungi. a Representative confocal microscope images of arbuscules formed by Ga165 (top) and Ri09 (bottom) at 3 (left) and 7 (right) weeks post-inoculation (WPI). Roots were stained with wheat germ agglutinin conjugated to Alexa Fluor 488. Scale bars = 50 µm. b–c Violin plots of arbuscular width at 3 (b) and 7 (c) weeks post-inoculation measured in roots colonized by Ga165 (grey) or Ri09 (dark grey) overlayed with box and whisker plots showing the median line, a box representing the interquartile range (IQR), and whiskers representing 1.5 times the IQR. Colonization was low at 3 weeks, so only 132 and 80 arbuscules were measured in Ga165- and Ri09-colonized roots, respectively; however, at 7 weeks, colonization was higher and 488 and 508 arbuscules were measured in Ga165- and Ri09-colonized roots, respectively. The mean arbuscule width for each fungal species at both time points was compared using a student’s t test (****p < 0.0001)
Although growth differences among the different treatments already were visible at 3 WPI (Supplemental Fig. S3), they became pronounced at 7 WPI (Fig. 2). Compared to non-mycorrhizal (NM) plants, Ri09 colonization resulted in greater shoot and root biomass, whereas Ga165 provided no growth benefits (Fig. 2a–b). Both fungi led to increased P content (Fig. 2c–d) and concentration (Supplemental Fig. S4) in the shoots and roots of their host, but Ri09 transferred more P than Ga165. Nitrogen-15 (15N) enrichment levels in the roots and shoots of NM plants were low (Fig. 2e–f), which indicates that there was no significant mass flow of 15N from the HC into the RC. The 15N enrichment levels in NM plants exceeded the natural abundance levels of 15N in the atmosphere (0.366%), but the 15N abundance in soils is higher than that in air, and we used in our experiments a substrate with 20% soil (Boddey et al. 2001; De Carvalho et al. 2017). Both fungi transferred significantly more 15N from the HC into the roots of their host, resulting in dramatically increased root 15N enrichment. However, the presence of 15N in the roots is not indicative of plant uptake as it is impossible to distinguish 15N that resides in the intraradical hyphae from 15N that has been released by the fungus and assimilated by the plant into root tissues. Thus, shoot 15N enrichment is a better measure of N transfer. For this parameter, only Ri09 resulted in a significant increase, suggesting that Ga165 only minimally increased the host plant’s access to N while Ri09 significantly increased it (Fig. 2f). Overall, these data indicate that the rate at which Ga165 and Ri09 colonize roots is different, but once they both achieve nearly complete colonization of the root system, only Ri09 colonization results in increased growth, most likely by providing the host plant with improved access to both P and N.

Biomass and nutrient content of Medicago truncatula plants colonized by different arbuscular mycorrhizal fungi. Shown are dry weight (a–b), phosphorus content (c–d), and (%) nitrogen-15 enrichment (e–f) of roots (left) and shoots (right) taken from plants grown for 7 weeks post-inoculation. The plants were inoculated with either Glomus aggregatum 165 (Ga165) or Rhizophagus irregularis 09 (Ri09) and compared to non-mycorrhizal (NM) control plants. Data points represent individual values for biological replicates (n = 5) and bars represent the mean of each treatment ± SEM. Different letters on the bars indicate statistically significant (p ≤ 0.05) differences based on one-way ANOVA and LSD test. F statistics and p values are shown in Supplemental Table S1
Colonization by either fungus alters gene expression more dramatically in the roots than in the shoots of Medicago truncatula
The primary aim of this study was to identify potential molecular mechanisms driving the observed increased growth and nutritional benefits of Ri09 compared to Ga165. Because arbuscules are the primary site of nutrient exchange and their density was comparable at 7 WPI, we only used roots from this time for RNA sequencing (RNA-seq) to evaluate the gene expression patterns in roots and shoots of mycorrhizal and non-mycorrhizal plants (Supplemental File 1). Analysis of the root data revealed distinct, treatment-specific clustering of biological replicates (n = 3, Ga165 and NM; n = 4, Ri09) using dendrograms, multidimensional scaling, and principal component analyses (Supplemental Fig. S5). In contrast, analysis of the shoot data showed no distinct clustering of biological replicates (n = 4, Ga165 and NM; n = 3, Ri09) within each treatment (Supplemental Fig. S6), suggesting that particularly in the roots, gene expression is driven by the fungal partner. Consequently, the overall number of significant differentially expressed genes (DEGs) was lower in the shoots than in the roots (Supplemental Fig. S7).
Both shared and distinct sets of genes are differentially regulated in Medicago truncatula by colonization with either fungus
To identify key differences in the response of the host plant to either fungal species, we sorted all DEGs from each treatment and used an arbitrary threshold of log2(fold-change) > 2 and a q-value < 0.05 (Fig. 3a). This threshold revealed that 364 genes were upregulated in roots colonized by either fungus, and 197 or 165 genes were upregulated by colonization with only Ga165 or Ri09, respectively (Fig. 3b). Ga165 and Ri09 colonization led to the downregulation of 41 shared genes, and 172 and 77 unique genes, respectively. The clustering of DEGs allowed us to identify specific groups of similarly expressed genes (Fig. 3c). Clusters A and B comprise genes that were strongly downregulated in mycorrhizal roots, cluster C1 contains genes that were upregulated in mycorrhizal roots, clusters C2 and D include genes primarily upregulated by Ga165 colonization, and cluster C3 contains genes exclusively upregulated by Ri09 colonization (Supplemental File 2).

Significantly differentially expressed genes in roots (a–c) and shoots (d–f) of Medicago truncatula colonized by different AM fungi. a, d Number of significantly differentially expressed genes (DEGs; log2[fold-change] > 2 and a q-value < 0.05) in plants colonized by Glomus aggregatum 165 (Ga165) or Rhizophagus irregularis 09 (Ri09) compared either to non-mycorrhizal (NM) control plants or to each other. b, e Venn diagrams of significantly upregulated (yellow) and downregulated (blue) DEGs unique to Ga165- and Ri09-colonized plants or shared by both. c, f Heat maps of fragments per kilobase of transcript per million mapped reads (FPKM) values + 1 for all significant DEGs from each treatment. Gene clusters are assigned based on second-order and sub-clusters based on third- or fourth-order branching. The genes represented in each cluster for panels c and f are not identical. Upregulated genes are shown in yellow and downregulated genes in blue with intensity based on row z-score. All DEGs in each plot for roots and shoots, respectively, are listed in Supplemental Files 2 and 3
Although fewer genes were differentially expressed in the shoots, many were above our threshold (Fig. 3d). We found that 48 and 14 genes were up, and 13 and 27 genes were downregulated in the shoots of Ga165- and Ri09-colonized plants, respectively (Fig. 3e). In contrast to the roots, clustering of DEGs based on expression patterns in the shoots (Fig. 3f) revealed that only a small number of genes were upregulated (cluster C1) or downregulated (cluster A) in mycorrhizal plants. In addition, some genes were only downregulated by Ga165 (cluster B) or by Ri09 colonization (cluster D), while others were primarily upregulated by Ga165 colonization (cluster C2; Supplemental File 3). Collectively, these results demonstrate that compared to shoots, gene expression patterns in roots respond more strongly to colonization with different AM fungi, and that both fungi lead to distinct differences particularly in the root and to a lesser degree also in the shoot transcriptome.
Genes critical for the establishment of AM symbiosis are more strongly upregulated by Rhizophagus irregularis 09 than Glomus aggregatum 165
We conducted a targeted analysis of the root RNA-seq data by first evaluating the expression of many genes known to play a role in AM symbiosis (Supplemental Table S2). Among six core strigolactone biosynthesis or exporter genes (Fig. 4a), we observed that two (DXS2 and D27) were upregulated in AM roots compared to the NM control, one (MAX1a) was only upregulated in Ri09-colonized roots, and three (CCD7, CCD8-1, and PDR1a) were not affected. Two GRAS transcription factors (NSP1 and NSP2) that regulate strigolactone biosynthesis and other symbiosis genes were also upregulated, particularly in Ri09-colonized roots. The combined increased expression of NSP1, NSP2, CCD7, and MAX1a in Ri09-colonized roots compared to Ga165-colonized roots suggests that Ri09 more strongly induces strigolactone biosynthesis than Ga165.

Expression patterns of genes encoding enzymes involved in the strigolactone (a) and gibberellic acid (b) biosynthesis pathways in the roots of Medicago truncatula colonized by arbuscular mycorrhizal fungi. Each diagram portrays the relative expression levels of genes that encode components of the pathways shown as fragments per kilobase of transcript per million mapped reads (FPKM; blue = low, yellow = high) in roots from non-mycorrhizal (NM) plants or plants colonized by either Glomus aggregatum 165 (Ga165) or Rhizophagus irregularis 09 (Ri09). Letters within boxes indicate significant differences (q-value < 0.05) in FPKM values between treatments and were determined as part of the CuffDiff2 analysis (see Supplemental Table S2). Abbreviations: GAP, glyceraldehyde 3‐phosphate; DXS2, 1‐deoxyxylulose 5‐phosphate synthase2; MEP, 2‐methyl‐erythritol‐4‐phosphate; GGDP geranylgeranyl pyrophosphate; D27, Dwarf27; CCD, carotenoid cleavage dioxygenase; and PDR1a, pleiotropic drug resistance 1a (a). CPS, ent-copalyl diphosphate synthase; KS, ent-kaurene synthase; KO, ent-kaurene oxidase; KAO, ent-kaurenoic acid oxidase; GA13ox, gibberellin 13-oxidase; GA20ox, gibberellin 20-oxidase; GA3ox, gibberellin 3-oxidase; and GA2ox, gibberellin 2-oxidase (b)
Among the CSSP genes, the lipochitooligosaccharide receptor NFP was downregulated and PUB1, a negative regulator of AM symbiosis (Vernié et al. 2016), was upregulated in roots colonized by either fungus (Supplemental Table S2). None of the other 13 analyzed CSSP genes was affected. Gibberellic acid (GA) is known to play an important role in regulating AM colonization (Floss et al. 2013; Nouri et al. 2021), and several genes regulating the biosynthesis and degradation of GA were differentially expressed by mycorrhizal colonization (Fig. 4b). In Ri09-colonized roots, the expression of the genes encoding ent-copalyl diphosphate synthase (CPS) and GA3oxidase (GA3ox) were downregulated compared to NM roots, but the gene encoding ent-kaurene oxidase (KO) was upregulated. In Ga165-colonized roots, four GA biosynthesis genes (GA13ox and three homologs of GA20ox) were significantly upregulated compared to NM roots. In Ri09-colonized roots, two of the GA20ox homologs were also upregulated, but not as strongly as by GA165. Similarly, the GA degradation gene GA2ox1 was upregulated in mycorrhizal roots, but most strongly in Ga165-colonized roots (Supplemental Table S2).
Given the higher expression of more GA biosynthesis genes in Ga165 compared to Ri09-colonized roots, we hypothesize that GA plays a stronger role in regulating the symbiosis with GA165 than with Ri09. Therefore, we evaluated the expression of genes downstream of the CSSP and DELLA1/2, which are proteins that are negatively regulated by GA (Davière and Achard 2013). We evaluated the expression of 21 genes (Supplemental Table S3), including seven AM-induced transcription factors that regulate arbuscule development, function, or degradation (DIP1, RAD1, RAM1, MIG1/2/3, and MYB1); three cellular remodeling genes (EXO70I, SYP132A, and VAPYRIN); two phosphate transporters (PT4 and PT8); three ammonium transporters (AMT2;3, AMT2;4, and AMT2;5); the putative lipid transporters STR and STR2 and three lipid biosynthesis genes (RAM2, FatM, and WRI5a); and the sugar transporter SWEET1.2. Almost all these genes were upregulated by both AM fungal species; however, six were substantially more upregulated by Ri09, including RAD1, MYB1, PT8, AMT2;3, RAM2, and SWEET1.2 (Fig. 5a–f). Together with the physiological results, the increased upregulation of PT8, AMT2;3, and SWEET1.2 in Ri09-colonized roots is suggestive of greater nutrient exchange activity across the PAM than that in Ga165-colonized roots. This is further supported by a suggestive positive relationship of both shoot P content versus PT8 expression (p = 0.09) and shoot 15N enrichment versus AMT2;3 expression (p = 0.08; Supplemental Fig. S8).

Relative expression of six genes in the roots of Medicago truncatula after colonization with different arbuscular mycorrhizal fungi. Expression of Required for Arbuscule Development 1 (RAD1) (a), MYB-like Transcription Factor 1 (MYB1) (b), Phosphate Transporter 8 (PT8) (c), Ammonium Transporter 2;3 (AMT2;3) (d); Reduced Arbuscular Mycorrhization 2 (RAM2) (e), and Sugars Will Eventually Be Exported Transporter 1.2 (SWEET1.2) (f) in the roots of non-mycorrhizal plants (NM, n = 3) and plants inoculated with Glomus aggregatum 165 (Ga165, n = 3) or Rhizophagus irregularis 09 (Ri09, n = 4). Expression levels are shown as fragments per kilobase of transcript per million mapped reads (FPKM). Data points represent individual values for biological replicates (n = 5) and bars represent the mean of each treatment ± SEM. Significant differences (q-value < 0.05) in FPKM values between treatments were determined as part of the CuffDiff2 analysis. Bars topped by the same letter do not differ significantly (see Supplemental Table S3)
Colonization with Glomus aggregatum 165 and Rhizophagus irregularis 09 differentially affects the expression of transporters
Gene ontology (GO) enrichment analysis revealed several cellular components, molecular functions, and/or biological processes that were significantly enriched among genes that are only upregulated (Supplemental Fig. S9) or downregulated (Supplemental Fig. S10) in AM roots. Predominant among these were GO terms related to transmembrane transport. We filtered all gene annotations containing the term “transport” from the M. truncatula JCVI 4.0v2 annotation file (Tang et al. 2014) and found 259 genes that were significantly differentially regulated in mycorrhizal roots (Supplemental File 5). Based on their function, we further subdivided these genes into two groups: transport of mineral nutrients (72 genes) and secondary metabolites (187 genes). Compared to NM roots, 12 mineral transporters were up- and 17 were downregulated by both AM fungi (Fig. 6a). This suggests that a core set of mineral transporters in AM roots are similarly regulated independent of fungal benefits. However, a unique set of transporters were differentially regulated by each AM fungal species: 14 were up- and 13 were downregulated in Ga165-colonized roots, while only 7 were up- and 12 were downregulated in Ri09-colonized roots. From the identified 16 groups of mineral transporters (Fig. 6b), we focused on ammonium, nitrate, phosphate, and sulfate transporters with FPKMs ≥ 10, due to the key role that AM fungi play in the transport of these nutrients (Fig. 6c–f). Among the five differentially regulated ammonium transporters, one belongs to the AMT1 ammonium transporter family and four to the AMT2 family. Compared to AM roots, AMT1;1 and two of the AMT2 family transporters (AMT2;1 and AMT2;6) were highly expressed in NM roots, while AMT2;3 and AMT2;5 were induced specifically in mycorrhizal roots. AMT2;3 was particularly upregulated in Ri09-colonized roots (Fig. 6c).

Expression patterns of significantly differentially expressed mineral transporter genes in the roots of Medicago truncatula after colonization with different arbuscular mycorrhizal fungi. a Venn diagrams showing the number of mineral transporters that are significantly up- (yellow) or downregulated (blue) in roots colonized by Glomus aggregatum 165 (Ga165) or Rhizophagus irregularis 09 (Ri09), or both. b Summary of the number of genes from specific gene annotation groups of mineral transporters that are up- or downregulated in roots colonized by Ga165 or Ri09, or both (see also Supplemental File 6). Expression patterns of genes annotated as ammonium (c), nitrate (d), phosphate (e), and sulfate (f) transporters in the roots of non-mycorrhizal plants (NM; n = 3) and plants inoculated with Ga165 (n = 3) or Ri09 (n = 4). The bars in c–f represent the mean of each treatment ± SEM (data points for biological replicates are displayed in Supplemental Fig. S11). Expression levels are shown as fragments per kilobase of transcript per million mapped reads (FPKM); only genes with FPKM ≥ 10 are shown; significant differences (q-value < 0.05) in FPKM values between treatments were determined as part of the CuffDiff2 analysis. Bars topped by the same letter do not differ significantly
We also identified seven differentially regulated nitrate transporters (Fig. 6d). Five belong to the nitrate/peptide transporter (NPF) family, and two are putative nitrate transporters. Interestingly, the most highly expressed nitrate transporter (NPF4.12) was highly upregulated in AM roots, particularly in Ri09-colonized roots. In contrast, five nitrate transporters were more highly expressed in Ga165-colonized roots than in Ri09-colonized roots (NPF1.2, NPF1.5, NPF1.6, Medtr4g114340, and Medtr1g057460), and two (NPF1.2 and Medtr1g057460) also differed from control roots. Only the nitrate transporters NPF5.24 and Medtr1g057460 showed higher expression in NM roots than in AM-colonized roots.
Seven phosphate transporters were differentially regulated (Fig. 6e). Four belong to the high-affinity inorganic phosphate transporter family PHT1 (PT4, PT6, PT8, and PT9), two were annotated as high-affinity inorganic phosphate transporters (Medtr1g043290 and Medtr1g074930), and one as a phosphate transporter PHO1-like protein. As expected, two of the PHT1 transporter genes, PT4 and PT8, were only expressed in AM roots, and PT8 expression was higher in Ri09-colonized roots. By contrast, the expression of Medtr1g074930 was strongly upregulated in Ga165-colonized roots compared to NM and Ri09-colonized roots. The PHO1-like transporter (Medtr1g041695) showed higher expression levels in NM than in AM roots.
We identified six sulfate transporters that were differentially regulated among the three treatments (Fig. 6f). Two were annotated as high-affinity sulfate transporter 1 (SULTR1) family genes (SULTR1.1 and 1.2), three as sulfate/bicarbonate/oxalate exchanger and transporter genes (SULTR2.2, 3.1, and 4.1), and one as a sulfate transporter-like gene (MOT1.1). While the expression levels of SULTR2.2 and SULTR4.1 were higher in NM roots than in AM roots, the remaining sulfate transporters were upregulated by colonization with Ri09 (SULTR1.1), Ga165 (SULTR3.1), or by both fungi (SULTR1.2 and MOT1.1).
In total, we identified 20 groups of secondary metabolite transporters in our dataset. Compared to NM control roots, 29 of these transporters were up- and 43 were downregulated in AM roots (Fig. 7a). Several secondary metabolite transporters were differentially regulated in Ga165- and Ri09-colonized roots. Among these, most belonged to six gene annotation groups, including ATP-binding cassette (ABC), amino acid, major facilitator superfamily (MFS), major intrinsic protein (MIP), peptide, and Sugars Will Eventually be Exported Transporters (SWEETs) (Fig. 7b). Here, we will only discuss those transporters that are known to play a role in AM symbiosis, or that were significantly differentially regulated by mycorrhizal colonization.

Expression patterns of significantly differentially expressed secondary metabolite transporters in the roots of Medicago truncatula after colonization with different AM fungi. a Venn diagrams showing the number of secondary metabolite transporters that are significantly up- (yellow) or downregulated (blue) in roots colonized by Glomus aggregatum 165 (Ga165) or Rhizophagus irregularis 09 (Ri09), or both. b Summary of the number of genes from specific gene annotation groups of secondary metabolite transports that are up- or downregulated in roots colonized by Ga165 or Ri09, or both (see Supplemental File 7). Expression patterns of genes annotated as ATP-binding cassette (ABC) (c), amino acid or peptide (d), major facilitator superfamily (MFS) or major intrinsic protein (MIP) (e), and SWEET transporters (f) in the roots of non-mycorrhizal plants (NM; n = 3) and plants inoculated with Ga165 (n = 3) or Ri09 (n = 4). The bars in c–f represent the mean of each treatment ± SEM (data points for biological replicates are displayed in Supplemental Fig. S12). Expression levels are shown as fragments per kilobase of transcript per million mapped reads (FPKM); only genes with FPKM ≥ 10 are shown; significant differences (q-value < 0.05) in FPKM values between treatments were determined as part of the CuffDiff2 analysis. Bars topped by the same letter do not differ significantly. Abbreviations in b: G3P, glycerol-3-phosphate; NAT, nucleobase-ascorbate transporter; OST, organic solute transporter; TPT, triose-phosphate transporter; TRAPP, transport protein particle
Two white-brown-complex ABC transporter family proteins (STR and STR2; Zhang et al. 2010), three ABC transporter B family proteins (AMN2, AMN3 [Roy et al. 2021], and Medtr4g124040), and one drug resistance transporter-like ABC domain protein (Medtr1g050525) were strongly upregulated in AM roots (Fig. 7c). There was a trend of higher expression levels in Ri09-colonized roots, but only the expression of AMN3 differed significantly from the expression in Ga165-colonized roots. The expression levels in Ga165-colonized roots were higher but only in the cases of STR2, AMN3, and Medtr1g050525 did they differ significantly from NM roots.
Two amino acid transporters, Medtr1g030660 and Medtr2g101920, and two peptide transporters, PTR3 and OPT15, were upregulated in mycorrhizal roots (Fig. 7d). As expected, NOPE1, an N-acetylglucosamine transporter of the major facilitator superfamily (MFS) that is required for AM symbiosis (Nadal et al. 2017), also was highly upregulated in AM roots (Fig. 7e). Three major intrinsic protein (MIP) transporters, NIP1;5, NIP1;6, and NIP3;1, which often are classified as aquaporins, were upregulated in mycorrhizal roots. NIP1;5 was particularly highly expressed in Ri09-colonized roots, while NIP3;1 and NIP6;2 showed higher expression levels in Ga165-colonized roots.
SWEETs are known to play a crucial role in AM symbiosis (Doidy et al. 2019), and five were differentially expressed in AM roots. SWEET1.2 was upregulated by colonization with either fungus, but especially with Ri09. SWEET3.3 and SWEET12 were only upregulated in Ri09-colonized roots, whereas SWEET7 and SWEET13 were only upregulated in Ga165-colonized roots (Fig. 7f). The variation in the expression patterns of these five SWEET genes suggests that sugar transport is strongly regulated in response to fungal benefit.
Photosynthesis-related genes in the shoots are upregulated in response to colonization with Rhizophagus irregularis 09
GO enrichment analysis on several gene clusters shown in Fig. 2f revealed that cellular components and biological processes associated with photosynthesis were enriched in the shoots of Ri09-colonized plants (Supplemental Fig. S13a). The associated genes were annotated as follows: Medtr1g115410, photosystem II reaction center family protein (orthologous to PsbP in Arabidopsis thaliana); Medtr2g082580, oxygen-evolving enhancer protein (orthologous to PsbQ-like protein 2 in A. thaliana); Medtr5g018670, photosystem II oxygen-evolving enhancer protein; Medtr1g015290, ultraviolet-B-repressible protein (orthologous to PsbX in A. thaliana); and Medtr1588s0010, ATP synthase F1, gamma subunit. The expression levels of PsbP, PsbQ-like protein 2, PsbX, and the ATP synthase gene were significantly higher in the shoots of Ri09-colonized plants than those in NM plants (Fig. 8a). Among these genes, only PsbP also was upregulated significantly in Ga165-colonized plants. By contrast, Medtr5g018670 was more highly expressed in the shoots of Ga165-colonized than in NM plants. Overall, the expression patterns of these photosynthesis-related genes followed a trend of low to high expression in NM, Ga165-colonized, and Ri09-colonized plants, respectively. This implies that photosynthesis was most strongly induced in the shoots of Ri09-colonized plants, which could explain the increased biomass of Ri09-colonized plants. However, whether this was a direct consequence of colonization by Ri09 or a side effect of its ability to provide the host plant with improved nutrient access cannot be determined from the data.

Expression patterns of significantly differentially expressed genes in the shoots of Medicago truncatula colonized by different AM fungi. Expression levels of genes involved in photosynthesis (a), response to biotic stress (b), and response to abiotic stress (c) in non-mycorrhizal plants (NM; n = 4) or plants colonized by Glomus aggregatum 165 (Ga165, n = 4) or Rhizophagus irregularis 09 (Ri09, n = 3). Bars represent the mean of each treatment ± SEM (data points for biological replicates are displayed in Supplemental Fig. S14). Expression levels are shown as fragments per kilobase of transcript per million mapped reads (FPKM), and significant differences (q-value < 0.05) in FPKM values between treatments were determined as part of the CuffDiff2 analysis. Bars topped by the same letter do not differ significantly (see also Supplemental Table S4)
Biotic and abiotic stress–related genes are differentially regulated in the shoots of Glomus aggregatum 165 and Rhizophagus irregularis 09 colonized plants
Many of the GO terms that were enriched in the shoots of Ga165-colonized plants encompassed biological processes related to biotic stress (Supplemental Fig. S13b). The genes linked to these GO terms were annotated as follows: Medtr4g054920, cytochrome P450 family 94 protein; Medtr4g120760, pathogenesis-related protein bet V I family protein; Medtr8g096900, pathogenesis-related thaumatin family protein; Medtr4g092010, (3S)-linalool/(E)-nerolidol/(E,E)-geranyl linalool synthase; and Medtr5g053950, allene oxide cyclase. Four of these five genes were significantly upregulated in Ga165-colonized plants compared to NM plants, while the expression of these genes in Ri09-colonized plants did not differ from NM plants (Fig. 8b). This could indicate that the host plant is responding to Ga165 as it would to a pathogen by upregulating defense responses. It also could indicate that Ga165 strongly stimulates host plant defenses against pathogens.
In contrast, genes downregulated by Ri09 colonization (Fig. 3f, cluster D) were associated with biotic and abiotic stress (Supplemental Fig. S13c). The genes included in these GO terms were annotated as follows: Medtr1g048990, superoxide dismutase; Medtr7g069980, ferritin; Medtr8g059170, NAC transcription factor–like protein; Medtr7g079180, late embryogenesis abundant protein; and Medtr3g087590, myo-inositol 1-phosphate synthase. The expression of Medtr7g079180 was significantly upregulated in the shoots of Ga165-colonized plants compared to NM plants, but the remaining abiotic stress response genes followed a trend of high, medium, and low expression in NM, Ga165-colonized, and Ri09-colonized plants, respectively (Fig. 8c). This expression pattern of abiotic stress response genes may indicate that M. truncatula experiences less abiotic stress during Ri09 than Ga165 colonization, possibly because of the improved supply of N and P to the host by Ri09.
Discussion
The growth and nutritional benefits of arbuscular mycorrhizal fungi are linked to mycorrhiza-induced transporter expression
When compatible host plants are inoculated with AM fungi, colonization most often leads to increased nutrient uptake and growth, especially under nutrient-deprived conditions (Chandrasekaran 2020). We found that under N and P limitation, Ri09 colonized M. truncatula faster than Ga165, but that after 7 WPI both fungi achieved equivalent colonization densities, including percent arbuscule root occupancy and arbuscule width (Fig. 1; Supplemental Fig. S2). Ri09 promoted greater growth benefits and provided more N and P to the host than Ga165 (Fig. 2). This observation is consistent with both the expression patterns that we observed for mycorrhiza-induced N and P transporters and with previous findings where we demonstrated that Ri09 is more cooperative than Ga165 (Kiers et al. 2011; Fellbaum et al. 2014; Wang et al. 2016). For example, the known AM-induced ammonium transporters AMT2;3 and AMT2;5 were upregulated by Ga165 and Ri09 (Fig. 6c). Although the expression levels of AMT2;3 in AM roots were lower than that of AMT2;5, AMT2;3 expression was more upregulated by Ri09 than by Ga165 colonization. This could suggest that the expression of these two genes with a similar function is regulated by different molecular mechanisms, which would allow a fine-tuning of ammonia uptake via the mycorrhizal pathway. Similar to AMT2;3, we observed the same expression pattern for the putative ammonium transporter NIP1;5 (Fig. 7e), which has not been functionally characterized in M. truncatula during AM symbiosis, although the soybean ortholog Nod26 plays a crucial role in ammonium transport in the rhizobia-legume symbiosis (Frare et al. 2018). Interestingly, although the AM fungi were provided with 15NH4Cl as an N source, we also found that the nitrate transporter NPF4.12 was strongly induced in AM roots, especially in Ri09-colonized roots (Fig. 6d). Aloui et al. (2018) found that NPF4.12 is enriched in the proteome of mycorrhizal M. truncatula roots, and recently the rice ortholog OsNPF4.5 was functionally characterized as an AM-induced nitrate transporter that is highly expressed in arbusculated cells and localizes to the plasma membrane (Wang et al. 2020).
Similar to the expression of AMT2;3, the AM-induced P transporters PT4 and PT8 were both highly expressed in AM-colonized roots, especially Ri09-colonized roots (Fig. 6e). Likewise, colonization with either fungus, but particularly with Ri09, caused an increase in shoot P content. We conducted a correlation analysis and found indications of a positive association of PT8 and AMT2;3 expression with shoot P content and shoot 15N enrichment, respectively (Supplemental Fig. S8). This together with the higher expression of these key mycorrhizal ammonium and nitrate transporters in Ri09- versus Ga165-colonized roots supports the view that PT8 and AMT2;3 play a role in the observed significant increase in shoot P content and 15N enrichment of Ri09-colonized plants.
Medtr1g074930, however, a putative high-affinity inorganic phosphate transporter, was strongly upregulated in Ga165-colonized roots (Fig. 6e). This P transporter is upregulated during P deficiency (Wang et al. 2017), and the upregulation of Medtr1g074930 in our study could indicate that the P transport by Ga165 was not sufficient to prevent P deficiency in the host plant. Notwithstanding, this is inconsistent with the observation that Medtr1g074930 was not upregulated in the roots of NM plants (Fig. 6e) and Ga165-colonized roots had a higher P concentration than NM roots (Supplemental Fig. S4). Regardless, others have similarly observed that the expression of high-affinity P transporters is differentially regulated in the host by colonization with different species of AM fungi (Grunwald et al. 2009).
Just as for N and P, AM fungi also play a critical role in supplying sulfur to the host plant (Sieh et al. 2013). We observed upregulation of two sulfate transporters, SULTR1.1 and SULTR1.2 (Fig. 6f). Both are upregulated in mycorrhizal M. truncatula roots, although not consistently (Casieri et al. 2012; Wipf et al. 2014). The SULTR1.2 ortholog in Lotus japonicus appears to play an important role in both direct and mycorrhizal sulfur uptake pathways, and its promoter is active in arbuscule-containing cells (Giovannetti et al. 2014). We also observed that the putative sulfate transporter gene MOT1.1 was upregulated in Ga165- and Ri09-colonized roots (Fig. 6f). However, the involvement of MOT1.1 in sulfur transport has not yet been experimentally demonstrated. MOT1.1 is phylogenetically related to MOT1.3, which is a molybdate transporter crucial for the rhizobia-legume symbiosis (Tejada-Jiménez et al. 2017).
Patterns of photosynthesis, carbon transporter, and lipid biosynthesis gene expression in the host plant are linked to the nutritional benefits provided by the fungal symbiont
In exchange for the nutritional benefits that AM fungi provide, plants transfer between 4 and 20% of their net fixed carbon in the form of carbohydrates and lipids to their fungal partners (Paul and Kucey 1981; Wright et al. 1998; Lendenmann et al. 2011; Roth and Paszkowski 2017). Both the nutritional benefits that AM fungi provide and the increased carbon sink they create are known to stimulate an increase in photosynthesis (Kaschuk et al. 2009). In line with this, we observed that the expression levels of a few photosynthesis-related genes were significantly upregulated in Ri09-colonized compared to NM plants, whereas expression in Ga165-colonized plants was intermediate (Fig. 8a). Thus, the elevated growth response provided by Ri09 (Fig. 2a–b) was likely not only because of increased nutrient transfer, but potentially also due to stimulation of photosynthesis.
Our previous studies have shown that host plants preferentially allocate more carbon to the more cooperative AM fungus Ri09 (Kiers et al. 2011). Yet, we found that the putative lipid transporters STR and STR2 were upregulated in AM roots but did not differ significantly between Ga165- and Ri09-colonized roots (Fig. 7c). Nevertheless, we did observe significant differences in the expression levels of the lipid biosynthesis gene RAM2 (Fig. 5e). It has been hypothesized that RAM2 is involved in the biosynthesis of βMAG (sn-2-monoacylglycerol), and that STR and STR2 export the 16:0 βMAG produced by RAM2 across the periarbuscular membrane to the AM fungus (Bravo et al. 2017). A higher biosynthesis of βMAG by RAM2 in Ri09-colonized roots could lead to increased lipid transfer to the AM fungus.
In addition, we found that the expression of the sugar transporter SWEET1.2 (Fig. 5f) was highest in Ri09-colonized roots, which strongly suggests an increase in transport of carbon resources to the fungus in exchange for elevated nutritional benefits (Bravo et al. 2017; An et al. 2019). Similar to our finding, others have found that SWEET1.2 is highly upregulated during AM colonization, particularly in arbusculated cortical cells, highlighting the critical role this sugar transporter plays in the AM symbiosis (An et al. 2019; Konečný et al. 2019). An et al. (2019), however, found that it is not essential for the maintenance of the AM symbiosis, most likely because of functional redundancy with other SWEETs. We found that SWEET3.3 and SWEET12 were more strongly upregulated in Ri09- than Ga165-colonized roots (Fig. 7f); both are upregulated in arbusculated cortical cells in M. truncatula (Sameeullah et al. 2016). Interestingly, SWEET7 and SWEET13 were strongly induced in Ga165-colonized roots, but SWEETs perform diverse physiological functions, and their upregulation may not necessarily increase sugar transport to the fungus. For example, some SWEET transporters in Arabidopsis localize to the tonoplast and restrict sugar efflux from the root (Chen et al. 2015). Given the variable expression patterns of these five SWEETs in Ri09- and Ga165-colonized roots and the different roles they may play in sugar efflux and retention, functionally characterizing them will improve our understanding of the mechanisms controlling carbon allocation during AM symbioses.
Differential expression of genes critical for the establishment of AM symbiosis likely contributes to the differential expression of nutrient transporters
To promote AM colonization, we grew the experimental plants under N and P limitation. It is known that P-deficient plants increase their production of strigolactones, which serve as signaling molecules that stimulate and attract AM fungi to colonize roots and thereby alleviate P deficiency (Yoneyama et al. 2007; Kafle et al. 2019). We observed in Ga165- and Ri09-colonized roots a similar expression of genes involved in the early steps of strigolactone biosynthesis (DXS2 and D27), but several downstream genes (CCD7, MAX1a, and both NSP1 and NSP2) were expressed at significantly higher levels in Ri09- than in Ga165-colonized roots. An overexpression of mycorrhiza-induced CLE peptides in M. truncatula that are involved in negatively regulating endogenous strigolactone biosynthesis caused both a decrease in strigolactone levels and AM colonization (Müller et al. 2019). This indicates a direct positive correlation between strigolactone levels and AM colonization. The high gene expression of strigolactone biosynthesis genes in Ri09-colonized roots could indicate that the strigolactone levels in Ri09-colonized roots were higher than those in Ga165-colonized roots (Fig. 4; Supplemental Table S2), but Ri09 and Ga165 reached similar levels of AM root colonization in our experiment. However, Müller et al. (2019) additionally showed that a knock-down of CLE53 and consequently diminished suppression of strigolactone biosynthesis resulted in higher PT4 expression. We also observed an increase in the expression of PT4 and other mycorrhiza-induced nutrient transporters in Ri09- versus Ga165-colonized roots. This could indicate that the increased expression of these nutrient transporters is linked to an increase in strigolactones.
While there were no significant differences in the expression of CSSP genes, we did observe a differential expression of key transcription factors in Ri09- and Ga165-colonized roots. For example, RAM1 (Park et al. 2015) and MIG2/3 (Heck et al. 2016) were both upregulated in Ga165- and Ri09-colonized roots. However, RAD1 (Park et al. 2015) and MYB1 (Floss et al. 2017), which are involved in arbuscule development and the suppression of arbuscule degeneration, respectively, were more highly induced in Ri09- compared to Ga165-colonized roots (Fig. 5a, b; Supplemental Table S3). Surprisingly, this did not result in measurable differences in arbuscule number or size (Fig. 1a), but it may have been a significant driver in altering the expression of nutrient transporters that localize to the periarbuscular membrane because transcription factors play a significant role in regulating this process (Pimprikar and Gutjahr 2018).
A recent study demonstrated that while GA is required for AM development, increasing concentrations of GA inhibit AM colonization and AM-related marker gene expression (Nouri et al. 2021). We observed that the GA biosynthetic gene KO was upregulated in Ri09-colonized roots compared to NM or Ga165-colonized roots, while several GA degrading genes (GA2ox1, GA13ox, and three homologs of GA20ox) were upregulated in Ga165-colonized roots compared to NM or Ri09-colonized roots. It has been hypothesized that GA biosynthesis is induced in AM roots, but that GA levels are maintained within narrow limits by GA breakdown and catabolism (Nouri et al. 2021). Given that the gene expression of the GA degrading enzymes GA2ox1 and GA3ox were higher in Ga165- than in Ri09-colonized roots, the endogenous GA levels in Ga165-colonized roots could have been lower than those in Ri09-colonized roots. Nouri et al. (2021) hypothesized that elevated GA levels cause degradation of DELLA proteins, and thereby lower the expression of AM-related genes, such as RAM1, RAM2, STR, and PT4, and that the expression of GA degradation and marker gene expression is controlled by the same mechanism. However, we found that the colonization levels of Ri09 and Ga165 did not differ at 7 WPI, and that the expression of RAM1, RAM2, and PT4 was particularly induced in Ri09-colonized roots. Together, this does not support the assumption that the GA levels in Ga165-colonized roots were lower than those in Ri09-colonized roots. Interestingly, exogenously supplied GA induced the expression of these marker genes (Nouri et al. 2021).
Arbuscular mycorrhiza–induced changes in the expression of host abiotic and biotic stress response genes are species specific
Although the primary focus of this study was to evaluate both resource exchange and transporter expression, our gene expression data also provided some unique insights into biotic and abiotic stress responses. Mycorrhizal growth responses are context-dependent and follow a mutualism to parasitism continuum (Johnson and Graham 2013). In natural environments, plants are colonized by communities of AM fungi, which supports the idea of functional complementarity, where a host plant experiences a broad range of benefits that are provided by specific members of the AM fungal community (e.g., enhanced nutrient availability or increased disease resistance; Jansa et al. 2008). While Ga165 does not seem to provide sufficient nutritional benefits to elicit a positive growth response, it is possible that like other AM fungal species it may provide resistance to one or more abiotic stresses (e.g., salinity or heavy metal accumulation), none of which was evaluated in this study.
We found that Ga165 more strongly upregulated defense-response genes in the shoots (Fig. 8b), two of which (Medtr4g120760 and Medtr8g096900) are upregulated in alfalfa when challenged with the foliar fungal pathogen Phoma medicaginis (Li et al. 2019a) and pea aphids (Acyrthosiphon pisum; Li et al. 2019b). There are two potential explanations for this observation. The plant may be exhibiting a similar response to a low-benefit AM fungus as it would to a fungal pathogen or Ga165 could be more effective than Ri09 at priming host defense responses and thereby prevent infection from pathogens and mitigate arthropod attack as previously has been described for other AM fungal species (Liu et al. 2007; Sharma et al. 2017).
Similarly, we also observed that genes associated with abiotic stress were upregulated in Ga165-colonized roots (Supplemental Fig. S9a). Three of these genes—Medtr5g063670, Medtr7g093170, and Medtr8g026960—were very strongly upregulated and are annotated as annexin D8, seed maturation protein, and homeobox-associated leucine zipper protein, respectively. Although the precise role of annexin D8 is not known, other annexins are differentially regulated during various abiotic and biotic stresses, and some seem to play a role during AM symbiosis (Roux et al. 2012). The seed maturation protein Medtr7g093170 has been characterized as the late embryogenesis abundant protein LEA1370, which is upregulated under various abiotic stresses, including abscisic acid treatment, cold, salinity, and dehydration (Zhang et al. 2020). Finally, the homeobox-associated leucine zipper protein encoded by Medtr8g026960 was characterized as a transcription factor that is upregulated during salt stress in alfalfa (Postnikova et al. 2013). However, because these genes are not yet functionally characterized, the question of whether Ga165 colonization could contribute to an alleviation of these abiotic stresses, or whether host plants respond similarly to abiotic stress and to Ga165 colonization, requires further studies.
It also is possible that Ga165 truly is a low-benefit symbiont capable of persisting in a community with high-benefit AM fungi, thereby avoiding both detection and subsequent sanctioning by the host plant for poor performance (Hart et al. 2013). Avoiding detection could be carried out using effectors, as has been described for the model AM fungus R. irregularis DAOM197198 which counteracts the plant immune program with the small secreted peptide SP7 (Kloppholz et al. 2011). Indeed, in our root expression data, we observed 11 defense response genes, including three defensins (Medtr8g012810, Medtr8g012815, and Medtr8g012845) were strongly upregulated in Ri09-colonized roots, but not in Ga165-colonized roots (Supplemental Fig. S9b; Supplemental Table S5). Defensins are known to inhibit pathogen growth (Maróti et al. 2015), so it is tempting to speculate that Ga165 may employ a suite of effectors to inhibit the expression of defensins and thereby temper host defense responses to avoid detection and sanctioning. Not all defensins play a role in pathogen response; some may play a role in the establishment of the AM symbiosis. For example, DefMd1 when overexpressed upregulates the expression of the GRAS transcription factor RAM1, which plays a critical role in arbuscule branching (Park et al. 2015; Uhe et al. 2018). In our study, we observed that RAM1 was highly upregulated in both Ga165- and Ri09-colonized roots (Supplemental Table S3). Further characterization of additional defensins is needed to determine their role in the establishment of the AM symbiosis.
Transporter expression patterns in Medicago truncatula roots reveal interesting candidates for future functional characterization
This study highlights the physiological and transcriptomic response of a host plant to colonization by both high- and low-benefit AM fungi, and Fig. 9 provides an overview of these responses. Our experimental approach allowed us to identify both the molecular mechanisms controlling the observed physiological responses and transporters that are both commonly and uniquely regulated by high-benefit (Ri09) and low-benefit (Ga165) AM fungal species. While the primary focus of our study was on N, P, and carbon transporters in the roots of M. truncatula colonized by Ga165 or Ri09, our analysis of differentially expressed transporters uncovered some interesting gene candidates with unique expression patterns (Figs. 6 and 7). Although there was a core set of transporters similarly induced by both fungi, a smaller set were expressed differently, and many are prime candidates for future functional analysis.

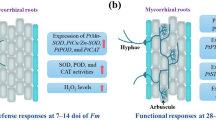
Summary of the physiological and transcriptomic responses in roots and shoots of Medicago truncatula colonized by either Glomus aggregatum 165 or Rhizophagus irregularis 09. Physiological data include the analysis of AM colonization, biomass, phosphorus (P) content, and % nitrogen-15 (15N) enrichment in plant tissues. The percent increase in each parameter relative to non-mycorrhizal control plants is shown. Transcriptomic data include the expression patterns of transporters and enrichment of gene ontology terms. Yellow up-arrows indicate an increase, equal signs (=) specify no significant change (p ≤ 0.05), and blue down-arrows designate a decrease in each response compared to non-mycorrhizal control plants. Arrow size indicates the degree of the response. *Despite the high average percent increase in 15N enrichment in the shoots of Ga165-colonized plants, the variability among replicates prevented the data from being considered significantly different from that of NM plants. †In the roots, it is impossible to distinguish 15N that still resides in intraradical hyphae and 15N that has been released by the fungus and assimilated into plant tissues; therefore, the reported significant increase in root % 15N enrichment is very likely an overestimate of the actual value
Multiple ABC transporters were induced by AM colonization (Fig. 7c), and two of these, STR and STR2, have already been studied extensively (Zhang et al. 2010). Two other transporters, AMN2 and AMN3, also are well studied, but the substrate they transport remains elusive (Roy et al. 2021). The two remaining genes, Medtr4g124040 and Medtr1g050525, have not been functionally characterized and could represent interesting targets for future studies. The amino acid transporters Medtr1g030660 and Medtr2g101920 and the peptide transporters PTR3 and OPT15 were upregulated in mycorrhizal roots. None of these has been functionally characterized, but the protein product of Medtr2g101920 was isolated from the plasma membrane of AM roots (Aloui et al. 2018). Future characterization of these transporters will provide insight into the extensive nutrient exchange mechanisms that support the AM symbiosis.
In conclusion, the comparison of the physiological and transcriptomic response of M. truncatula to colonization by two AM fungal species that provide different nutritional benefits to the host allowed us to identify potential molecular mechanisms that control reciprocal nutrient exchange. Top gene candidates include specific N and P transporters that already have been functionally characterized (e.g., AMT2;3, AMT2;5, NPF4.12, PT4, and PT8) and others that have not (e.g., NIP1;5). In addition, this study uncovered differential expression of genes putatively involved in the allocation of carbon resources to AM fungi depending on their nutritional benefit (e.g., RAM2, SWEET1.2, SWEET3.3, and SWEET12 which were upregulated in Ri09-colonized roots, while SWEET7 and SWEET13 were upregulated in Ga165-colonized roots).
Finally, this study highlights that root colonization with diverse fungal communities comprising both low- and high-benefit fungi could allow the host plant to not only choose among multiple trading partners, but also take advantage of the diverse benefits that fungi within that community can provide. The differential gene expression in the roots and shoots after colonization with AM fungi that differ in the symbiotic benefit that they provide indicates that host plants can fine tune their response to different fungi in their community.
Prior to this study, others have shown that host plants can distinguish between different AM fungal species, including when two fungi are colonizing individual root halves of a single plant in split root systems or when two fungal species are co-colonizing the same root system (Kiers et al. 2011; Zheng et al. 2015; Kafle et al. 2022). We argue that this selection can occur at the cellular level as gene expression within a single cell can be regulated based on the nutrient status of an individual cortical cell. The tools to directly test this idea are beginning to emerge (e.g., FRET sensors that can detect individual cortical cell P contents; Zhang et al. 2022), but some have yet to be developed (e.g., the ability to transform individual AM fungi with fluorescent reporters to allow for distinguishing two species co-colonizing the same root, as has been done with rhizobia; Stuurman et al. 2007). Thus, we anticipate the future ability of observing host plant sanctioning of low-benefit fungi and the rewarding of high-benefit fungi at the cellular level in roots co-colonized by more than one AM fungal strain.
Data and material availability
The RNA-seq datasets generated for and analyzed in the current study are available on NCBI as BioProject ID PRJNA832760 and can be accessed at http://www.ncbi.nlm.nih.gov/bioproject/832760.
References
Aloui A, Recorbet G, Lemaître-Guillier C et al (2018) The plasma membrane proteome of Medicago truncatula roots as modified by arbuscular mycorrhizal symbiosis. Mycorrhiza 28:1–16. https://doi.org/10.1007/s00572-017-0789-5
An J, Zeng T, Ji C et al (2019) A Medicago truncatula SWEET transporter implicated in arbuscule maintenance during arbuscular mycorrhizal symbiosis. New Phytol 224:396–408. https://doi.org/10.1111/nph.15975
Besserer A, Becard G, Jauneau A et al (2008) GR24, a synthetic analog of strigolactones, stimulates the mitosis and growth of the abuscular mycorrhizal fungus Gigaspora rosea by boosting its energy metabolism. Plant Physiol 148:402–413. https://doi.org/10.1104/pp.108.121400
Boddey RM, Polidoro JC, Resende AS et al (2001) Use of the 15N natural abundance technique for the quantification of the contribution of N2 fixation to sugar cane and other grasses. Aust J Plant Physiol 28:889–895. https://doi.org/10.1071/PP01058
Bravo A, Brands M, Wewer V et al (2017) Arbuscular mycorrhiza-specific enzymes FatM and RAM2 fine-tune lipid biosynthesis to promote development of arbuscular mycorrhiza. New Phytol 214:1631–1645. https://doi.org/10.1111/nph.14533
Breuillin-Sessoms F, Floss DS, Karen Gomez S et al (2015) Suppression of arbuscule degeneration in Medicago truncatula phosphate transporter4 mutants is dependent on the ammonium transporter 2 family protein AMT2;3. Plant Cell 27:1352–1366. https://doi.org/10.1105/tpc.114.131144
Brundrett MC, Tedersoo L (2018) Evolutionary history of mycorrhizal symbioses and global host plant diversity. New Phytol 220:1108–1115. https://doi.org/10.1111/nph.14976
Bücking H, Kafle A (2015) Role of arbuscular mycorrhizal fungi in the nitrogen uptake of plants: current knowledge and research gaps. Agronomy 5:587–612. https://doi.org/10.3390/agronomy5040587
Casieri L, Gallardo K, Wipf D (2012) Transcriptional response of Medicago truncatula sulphate transporters to arbuscular mycorrhizal symbiosis with and without sulphur stress. Planta 235:1431–1447. https://doi.org/10.1007/s00425-012-1645-7
Chandrasekaran M (2020) A meta-analytical approach on arbuscular mycorrhizal fungi inoculation efficiency on plant growth and nutrient uptake. Agriculture 10:370. https://doi.org/10.3390/agriculture10090370
Chen HY, Huh JH, Yu YC et al (2015) The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J 83:1046–1058. https://doi.org/10.1111/tpj.12948
Cope KR, Bascaules A, Irving TB et al (2019) The ectomycorrhizal fungus Laccaria bicolor produces lipochitooligosaccharides and uses the common symbiosis pathway to colonize populus roots. Plant Cell 31:2386–2410. https://doi.org/10.1105/tpc.18.00676
Davière JM, Achard P (2013) Gibberellin Signaling in Plants Development 140:1147–1151. https://doi.org/10.1242/dev.087650
De Carvalho EX, Menezes RSC, Santiago de Freitas AD et al (2017) The 15N natural abundance technique to assess the potential of biological nitrogen fixation (BNF) in some important C4 grasses. AJCS 11:1559–1564. https://doi.org/10.21475/ajcs.17.11.12.pne729
Doidy J, Vidal U, Lemoine R (2019) Sugar transporters in Fabaceae, featuring SUT MST and SWEET families of the model plant Medicago truncatula and the agricultural crop Pisum sativum. PLoS One 14:e0223173. https://doi.org/10.1371/journal.pone.0223173
Fellbaum CR, Gachomo EW, Beesetty Y et al (2012) Carbon availability triggers fungal nitrogen uptake and transport in arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci 109:2666–2671. https://doi.org/10.1073/pnas.1118650109
Fellbaum CR, Mensah JA, Cloos AJ et al (2014) Fungal nutrient allocation in common mycorrhizal networks is regulated by the carbon source strength of individual host plants. New Phytol 203:646–656. https://doi.org/10.1111/nph.12827
Feng F, Sun J, Radhakrishnan GV et al (2019) A combination of chitooligosaccharide and lipochitooligosaccharide recognition promotes arbuscular mycorrhizal associations in Medicago truncatula. Nat Commun 10:1–12. https://doi.org/10.1038/s41467-019-12999-5
Floss DS, Gomez SK, Park HJ et al (2017) A transcriptional program for arbuscule degeneration during AM symbiosis is regulated by MYB1. Curr Biol 27:1206–1212. https://doi.org/10.1016/j.cub.2017.03.003
Floss DS, Levy JG, Lévesque-Tremblay V et al (2013) DELLA proteins regulate arbuscule formation in arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci U S A 110:E5025–E5034. https://doi.org/10.1073/pnas.1308973110
Frare R, Ayub N, Alleva K, Soto G (2018) The ammonium channel NOD26 is the evolutionary innovation that drives the emergence, consolidation, and dissemination of nitrogen-fixing symbiosis in angiosperms. J Mol Evol 86:554–565. https://doi.org/10.1007/s00239-018-9867-3
Garcia K, Doidy J, Zimmermann SD et al (2016) Take a trip through the plant and fungal transportome of mycorrhiza. Trends Plant Sci 21:937–950
Genre A, Chabaud M, Balzergue C et al (2013) Short-chain chitin oligomers from arbuscular mycorrhizal fungi trigger nuclear Ca2+ spiking in Medicago truncatula roots and their production is enhanced by strigolactone. New Phytol 198:190–202. https://doi.org/10.1111/nph.12146
Giovannetti M, Tolosano M, Volpe V et al (2014) Identification and functional characterization of a sulfate transporter induced by both sulfur starvation and mycorrhiza formation in Lotus japonicus. New Phytol 204:609–619. https://doi.org/10.1111/nph.12949
Goff LA, Trapnell C, Kelley D (2012) CummeRbund: visualization and exploration of Cufflinks high-throughput sequencing data. R Packag Version 2:1–45
Grunwald U, Guo W, Fischer K et al (2009) Overlapping expression patterns and differential transcript levels of phosphate transporter genes in arbuscular mycorrhizal, Pi-fertilised and phytohormone-treated Medicago truncatula roots. Planta 229:1023–1034. https://doi.org/10.1007/s00425-008-0877-z
Hart MM, Forsythe J, Oshowski B et al (2013) Hiding in a crowd - does diversity facilitate persistence of a low-quality fungal partner in the mycorrhizal symbiosis? Symbiosis 59:47–56. https://doi.org/10.1007/s13199-012-0197-8
Havlin JL, Beaton JD, Tisdale SL, Nelson WL (2014) Soil fertility and fertilizers: an introduction to nutrient management. Pearson Education Inc, Eighth
Heck C, Kuhn H, Heidt S et al (2016) Symbiotic fungi control plant root cortex development through the novel GRAS transcription factor MIG1. Curr Biol 26:2770–2778. https://doi.org/10.1016/j.cub.2016.07.059
Horváth B, Yeun LH, Domonkos Á et al (2011) Medicago truncatula IPD3 is a member of the common symbiotic signaling pathway required for rhizobial and mycorrhizal symbioses. Mol Plant-Microbe Interact 24:1345–1358. https://doi.org/10.1094/mpmi-01-11-0015
Ingestad T (1960) Studies on the nutrition of forest tree seedlings. III mineral nutrition of pine. Physiol Plant 13:513–533. https://doi.org/10.1111/j.1399-3054.1960.tb08074.x
Jansa J, Smith FA, Smith SE (2008) Are there benefits of simultaneous root colonization by different arbuscular mycorrhizal fungi? New Phytol 177:779–789. https://doi.org/10.1111/j.1469-8137.2007.02294.x
Javot H, Penmetsa RV, Terzaghi N et al (2007) A Medicago truncatula phosphate transporter indispensable for the arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci 104:1720–1725. https://doi.org/10.1073/pnas.0608136104
Johnson NC, Graham JH (2013) The continuum concept remains a useful framework for studying mycorrhizal functioning. Plant Soil 363:411–419. https://doi.org/10.1007/s11104-012-1406-1
Kafle A, Cope KR, Raths R et al (2019) Harnessing soil microbes to improve plant phosphate efficiency in cropping systems. Agronomy 9:127. https://doi.org/10.3390/agronomy9030127
Kafle A, Frank HER, Rose BD, Garcia K (2022) Split down the middle: studying arbuscular mycorrhizal and ectomycorrhizal symbioses using split-root assays. J Exp Bot 73:1288–1300. https://doi.org/10.1093/JXB/ERAB489
Kafle A, Garcia K, Wang X et al (2018) Nutrient demand and fungal access to resources control the carbon allocation to the symbiotic partners in tripartite interactions of Medicago truncatula. Plant Cell Environ 1–15. https://doi.org/10.1111/pce.13359
Kaschuk G, Kuyper TW, Leffelaar PA et al (2009) Are the rates of photosynthesis stimulated by the carbon sink strength of rhizobial and arbuscular mycorrhizal symbioses? Soil Biol Biochem 41:1233–1244. https://doi.org/10.1016/j.soilbio.2009.03.005
Kiers ET, Duhamel M, Beesetty Y et al (2011) Reciprocal rewards stabilize cooperation in the mycorrhizal symbiosis. Science (80- ) 333:880–882. https://doi.org/10.1126/science.1208473
Kim D, Paggi JM, Park C et al (2019a) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. https://doi.org/10.1038/s41587-019-0201-4
Kim S, Zeng W, Bernard S et al (2019b) Ca2+-regulated Ca2+ channels with an RCK gating ring control plant symbiotic associations. Nat Commun 10:1–12. https://doi.org/10.1038/s41467-019-11698-5
Kloppholz S, Kuhn H, Requena N (2011) A secreted fungal effector of Glomus intraradices promotes symbiotic biotrophy. Curr Biol 21:1204–1209. https://doi.org/10.1016/j.cub.2011.06.044
Konečný J, Hršelová H, Bukovská P et al (2019) Correlative evidence for co-regulation of phosphorus and carbon exchanges with symbiotic fungus in the arbuscular mycorrhizal Medicago truncatula. PLoS One 14. https://doi.org/10.1371/journal.pone.0224938
Kretzschmar T, Kohlen W, Sasse J et al (2012) A petunia ABC protein controls strigolactone-dependent symbiotic signalling and branching. Nature 483:341–344. https://doi.org/10.1038/nature10873
LeBauer DS, Treseder KK (2008) Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology 89:371–379. https://doi.org/10.1890/06-2057.1
Lendenmann M, Thonar C, Barnard RL et al (2011) Symbiont identity matters: carbon and phosphorus fluxes between Medicago truncatula and different arbuscular mycorrhizal fungi. Mycorrhiza 21:689–702. https://doi.org/10.1007/S00572-011-0371-5/FIGURES/4
Lévy J, Bres C, Geurts R et al (2004) A putative Ca2+ and calmodulin-dependent protein kinase required for bacterial and fungal symbioses. Science (80- ) 303:1361–4. https://doi.org/10.1126/science.1093038
Li Y, Duan T, Nan Z, Li Y (2019a) Arbuscular mycorrhizal fungus alleviates alfalfa leaf spots caused by Phoma medicaginis revealed by RNA-seq analysis. J Appl Microbiol. https://doi.org/10.1111/jam.14387
Li Y, Nan Z, Duan T (2019b) Rhizophagus intraradices promotes alfalfa (Medicago sativaM) defense against pea aphids (Acyrthosiphon pisum) revealed by RNA-Seq analysis. Mycorrhiza 29:623–635. https://doi.org/10.1007/s00572-019-00915-0
Liu J, Maldonado-Mendoza I, Lopez-Meyer M et al (2007) Arbuscular mycorrhizal symbiosis is accompanied by local and systemic alterations in gene expression and an increase in disease resistance in the shoots. Plant J 50:529–544. https://doi.org/10.1111/j.1365-313X.2007.03069.x
MacLean AM, Bravo A, Harrison MJ (2017) Plant signaling and metabolic pathways enabling arbuscular mycorrhizal symbiosis. Plant Cell 29:2319–2335. https://doi.org/10.1105/tpc.17.00555
Maillet F, Poinsot V, André O et al (2011) Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature 469:58–64. https://doi.org/10.1038/nature09622
Maróti G, Downie JA, Kondorosi É (2015) Plant cysteine-rich peptides that inhibit pathogen growth and control rhizobial differentiation in legume nodules. Curr Opin Plant Biol 26:57–63. https://doi.org/10.1016/j.pbi.2015.05.031
Martin FM, Uroz S, Barker DG (2017) Ancestral alliances: plant mutualistic symbioses with fungi and bacteria. Science (80-) 356:eaad4501
McGonigle TP, Miller MH, Evans DG et al (1990) A new method which gives an objective measure of colonization of roots by vesicular-arbuscular mycorrhizal fungi. New Phytol 115:495–501. https://doi.org/10.1111/j.1469-8137.1990.tb00476.x
Müller LM, Flokova K, Schnabel E et al (2019) A CLE–SUNN module regulates strigolactone content and fungal colonization in arbuscular mycorrhiza. Nat Plants 5:933–939. https://doi.org/10.1038/s41477-019-0501-1
Nadal M, Sawers R, Naseem S et al (2017) An N-acetylglucosamine transporter required for arbuscular mycorrhizal symbioses in rice and maize. Nat Plants 3:1–7. https://doi.org/10.1038/nplants.2017.73
Nouri E, Surve R, Bapaume L et al (2021) Phosphate suppression of arbuscular mycorrhizal symbiosis involves gibberellic acid signaling. Plant Cell Physiol. https://doi.org/10.1093/pcp/pcab063
Park HJ, Floss DS, Levesque-Tremblay V et al (2015) Hyphal branching during arbuscule development requires RAM1. Plant Physiol 169. https://doi.org/10.1104/pp.15.01155
Paul EA, Kucey RMN (1981) Carbon flow in plant microbial associations incriminated in perinatal morbidity and mortality. Science (80-) 213:473–474. https://doi.org/10.1126/science.213.4506.473
Pertea M, Pertea GM, Antonescu CM et al (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295. https://doi.org/10.1038/nbt.3122
Pimprikar P, Carbonnel S, Paries M et al (2016) A CCaMK-CYCLOPS-DELLA complex activates transcription of RAM1 to regulate arbuscule branching. Curr Biol 26:987–998. https://doi.org/10.1016/j.cub.2016.01.069
Pimprikar P, Gutjahr C (2018) Transcriptional regulation of arbuscular mycorrhiza development. Plant Cell Physiol 59:673–690
Postnikova OA, Shao J, Nemchinov LG (2013) Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol 54:1041–1055. https://doi.org/10.1093/pcp/pct056
Roth R, Paszkowski U (2017) Plant carbon nourishment of arbuscular mycorrhizal fungi. Curr Opin Plant Biol 39:50–56. https://doi.org/10.1016/j.pbi.2017.05.008
Roux SJ, Clark GB, Morgan RO et al (2012) Evolutionary adaptation of plant annexins has diversified their molecular structures, interactions and functional roles. New Phytol 196:695–712. https://doi.org/10.1111/j.1469-8137.2012.04308.x
Roy S, Breakspear A, Cousins D et al (2021) Three common symbiotic ABC-B transporters in Medicago truncatula are regulated by a NIN-independent branch of the symbiosis signalling pathway. Mol Plant-Microbe Interact. https://doi.org/10.1094/mpmi-02-21-0036-r
Rush TA, Puech-Pagès V, Bascaules A et al (2020) Lipo-chitooligosaccharides as regulatory signals of fungal growth and development. Nat Commun 11:1–10. https://doi.org/10.1038/s41467-020-17615-5
Sameeullah M, Demiral T, Aslam N et al (2016) In silico functional analyses of SWEETs reveal cues for their role in AMF symbiosis. In: Plant, soil and microbes: volume 2: mechanisms and molecular interactions. Springer International Publishing, pp 45–58
Schindelin J, Arganda-Carreras I, Frise E et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Sharma E, Anand G, Kapoor R (2017) Terpenoids in plant and arbuscular mycorrhiza-reinforced defence against herbivorous insects. Ann Bot 119:791–801
Sieh D, Watanabe M, Devers EA et al (2013) The arbuscular mycorrhizal symbiosis influences sulfur starvation responses of Medicago truncatula. New Phytol 197:606–616. https://doi.org/10.1111/nph.12034
Singh S, Parniske M (2012) Activation of calcium- and calmodulin-dependent protein kinase (CCaMK), the central regulator of plant root endosymbiosis. Curr Opin Plant Biol 15:444–453
Spatafora JW, Chang Y, Benny GL et al (2016) A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108:1028–1046. https://doi.org/10.3852/16-042
St-Arnaud M, Hamel C, Vimard B et al (1996) Enhanced hyphal growth and spore production of the arbuscular mycorrhizal fungus Glomus intraradices in an in vitro system in the absence of host roots. Mycol Res 100:328–332. https://doi.org/10.1016/S0953-7562(96)80164-X
Stracke S, Kistner C, Yoshida S et al (2002) A plant receptor-like kinase required for both bacterial and fungal symbiosis. Nature 417:959–962. https://doi.org/10.1038/nature00841
Stuurman N, Pacios Bras C, Schlaman HRM et al (2007) Use of green fluorescent protein color variants expressed on stable broad-host-range vectors to visualize rhizobia interacting with plants. 13:1163–1169. https://doi.org/10.1094/MPMI.2000.13.11.1163
Tang H, Krishnakumar V, Bidwell S et al (2014) An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15:312. https://doi.org/10.1186/1471-2164-15-312
Tejada-Jiménez M, Gil-Díez P, León-Mediavilla J et al (2017) Medicago truncatula molybdate transporter type 1 (MtMOT1.3) is a plasma membrane molybdenum transporter required for nitrogenase activity in root nodules under molybdenum deficiency. New Phytol 216:1223–1235. https://doi.org/10.1111/nph.14739
The R Foundation (2020) R: the R project for statistical computing. https://www.r-project.org/. Accessed 11 May 2020
Trapnell C, Hendrickson DG, Sauvageau M et al (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31:46–53. https://doi.org/10.1038/nbt.2450
Uhe M, Hogekamp C, Hartmann RM et al (2018) The mycorrhiza-dependent defensin MtDefMd1 of Medicago truncatula acts during the late restructuring stages of arbusculecontaining cells. PLoS One 13:e0191841. https://doi.org/10.1371/journal.pone.0191841
Venkateshwaran M, Jayaraman D, Chabaud M et al (2015) A role for the mevalonate pathway in early plant symbiotic signaling. Proc Natl Acad Sci 112:9781–9786. https://doi.org/10.1073/pnas.1413762112
Vernié T, Camut S, Camps C et al (2016) PUB1 interacts with the receptor kinase DMI2 and negatively regulates rhizobial and arbuscular mycorrhizal symbioses through its ubiquitination activity in Medicago truncatula. Plant Physiol 170:2312–2324. https://doi.org/10.1104/pp.15.01694
Vierheilig H, Coughlan AP, Wyss U, Piché Y (1998) Ink and vinegar, a simple staining technique for arbuscular-mycorrhizal fungi. Appl Environ Microbiol 64:5004–5007. https://doi.org/10.1128/aem.64.12.5004-5007.1998
Vitousek PM, Porder S, Houlton BZ, Chadwick OA (2010) Terrestrial phosphorus limitation: mechanisms, implications, and nitrogen–phosphorus interactions. Ecol Appl 20:5–15. https://doi.org/10.1890/08-0127.1
Wang S, Chen A, Xie K et al (2020) Functional analysis of the OsNPF4.5 nitrate transporter reveals a conserved mycorrhizal pathway of nitrogen acquisition in plants. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2000926117
Wang T, Zhao M, Zhang X et al (2017) Novel phosphate deficiency-responsive long non-coding RNAs in the legume model plant Medicago truncatula. J Exp Bot 68:5937–5948. https://doi.org/10.1093/jxb/erx384
Wang X, Zhao S, Bücking H (2016) Arbuscular mycorrhizal growth responses are fungal specific but do not differ between soybean genotypes with different phosphate efficiency. Ann Bot 118:11–21. https://doi.org/10.1093/aob/mcw074
Wipf D, Mongelard G, van Tuinen D et al (2014) Transcriptional responses of Medicago truncatula upon sulfur deficiency stress and arbuscular mycorrhizal symbiosis. Front Plant Sci 5:680. https://doi.org/10.3389/fpls.2014.00680
Wright DP, Read DJ, Scholes JD (1998) Mycorrhizal sink strength influences whole plant carbon balance of Trifolium repens L. Plant, Cell Environ 21:881–891. https://doi.org/10.1046/j.1365-3040.1998.00351.x
Yoneyama K, Yoneyama K, Takeuchi Y, Sekimoto H (2007) Phosphorus deficiency in red clover promotes exudation of orobanchol, the signal for mycorrhizal symbionts and germination stimulant for root parasites. Planta 225:1031–1038. https://doi.org/10.1007/s00425-006-0410-1
Zhang Q, Blaylock LA, Harrison MJ (2010) Two Medicago truncatula half-ABC transporters are essential for arbuscule development in arbuscular mycorrhizal symbiosis. Plant Cell 22:1483–1497. https://doi.org/10.1105/tpc.110.074955
Zhang S, Daniels DA, Ivanov S et al (2022) A genetically encoded biosensor reveals spatiotemporal variation in cellular phosphate content in Brachypodium distachyon mycorrhizal roots. New Phytol. https://doi.org/10.1111/NPH.18081
Zhang YM, Wang HQ, Liu DM, Liu RJ (2020) Three tandemly aligned lea genes from Medicago truncatula confer differential protection to Escherichia coli against abiotic stresses. Biol Plant 64:95–103. https://doi.org/10.32615/bp.2019.112
Zheng C, Ji B, Zhang J et al (2015) Shading decreases plant carbon preferential allocation towards the most beneficial mycorrhizal mutualist. New Phytol 205:361–368. https://doi.org/10.1111/nph.13025
Acknowledgements
We would like to thank Lindsay McKeever for performing the Kjeldahl degradations for the 15N/14N analyses.
Funding
This work was funded by the USDA (2017–67014-26530), the SD Soybean Research and Promotion Council, and the Agricultural Experiment Station at SDSU to HB and SS. KG acknowledges the financial support of the North Carolina Agriculture Research Service (NCARS), the North Carolina Soybean Producers Association (2019–1656), and the USDA (2020–67013-31800).
Author information
Authors and Affiliations
Contributions
HB and SS conceived the original research plans; HB and AK designed the experiments; AK and JY performed the experiments, KRC and KG conducted the transcriptome analysis, and PP and GS completed the stable isotope analysis; KRC, AK, KG, JY, and PP analyzed the data; KRC wrote the article with input from all the authors; HB and SS supervised and completed the writing. HB agrees to serve as the author responsible for contact and ensures communication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cope, K.R., Kafle, A., Yakha, J.K. et al. Physiological and transcriptomic response of Medicago truncatula to colonization by high- or low-benefit arbuscular mycorrhizal fungi. Mycorrhiza 32, 281–303 (2022). https://doi.org/10.1007/s00572-022-01077-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00572-022-01077-2