
Abstract
Approximately 1% of all patients with Sjögren’s syndrome (SS) are children. Unlike the adult form, in which sicca syndrome is the main presentation, in children, the most common clinical finding is recurrent enlargement of the salivary glands. In pediatric SS, extraglandular manifestations represent a significant feature and, among these, kidney manifestations are relevant. Kidney involvement is observed in 5–20.5% of children with SS, most frequently tubulointerstitial nephritis. This injury can lead to serious phenotypes, including distal kidney tubular acidosis with the development of severe hypokalemia, which can lead to ECG abnormalities, weakness, and hypokalemic periodic paralysis. Kidney implications in pediatric SS also include nephrolithiasis, nephrocalcinosis, and various types of glomerular damage, which often require immunosuppressive therapies. Laboratory findings are usually comparable to adults, including hyperglobulinemia and high rates of antinuclear antibodies (ANA, 63.6–96.2%), and anti-Ro/SSA (36.4–84.6%). The current classification criteria for SS are inaccurate for the pediatric population, and more specific criteria are needed to improve the diagnostic rate. Due to the rarity of the disease, strong recommendations for treatment are lacking, and several therapeutic strategies have been reported, mostly based on glucocorticoids and disease-modifying antirheumatic drugs, with different outcomes. The aim of this paper is to provide an overview of the kidney implications of pediatric SS based on the latest evidence of the medical literature.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sjögren’s syndrome (SS) is a chronic autoimmune disorder that predominantly affects the exocrine glands, with frequent multi-organ involvement. In the adult population, the inflammation of salivary and lacrimal glands represents the main feature of the disease, leading to the development of sicca syndrome with xerostomia and xerophthalmia, resulting in dry mouth and eyes. Two forms of SS can be distinguished: primary SS (pSS) is recognized as an isolated disease, whereas secondary SS is associated with other underlying autoimmune disorders, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and systemic sclerosis [1]. The pathophysiology of SS is not entirely understood. Host and environmental factors lead to abnormal activation of both innate and adaptive immunity in genetically predisposed individuals [2, 3]. Lymphocytes and specific autoantibodies all contribute to the pathogenesis of chronic inflammation, leading to the formation of germinal centers in affected tissue and an increased risk of non-Hodgkin’s lymphoma and other malignancies [2,3,4]. Although SS is commonly observed in adults, mainly due to secondary forms, a childhood onset can rarely occur [2, 3, 5,6,7,8,9,10]. Indeed, pediatric SS is believed to account for approximately 1% of all SS patients and, 10 years ago, less than 200 cases of pediatric SS had been described [6, 11,12,13,14,15,16]. Typical clinical findings in children with SS include recurrent parotid swelling and, often, significant extraglandular manifestations (EGMs) (Fig. 1) [16, 17]. Due to the systemic spectrum of the disease, autoinflammation can affect every tissue and organ, resulting in a clinical phenotype that is highly heterogeneous [15, 17]. Kidney involvement is regarded as a relevant feature in pediatric SS, with an estimated prevalence of 5–20.5% (Table 1) [2, 3, 7,8,9,10, 16,17,18,19,20,21]. However, due to the disease’s rarity, epidemiological and clinical data regarding the kidney manifestations of pediatric SS are limited [6, 7, 17]. The most common kidney injury in affected children is tubulointerstitial nephritis (TIN), which can clinically manifest as renal tubular acidosis (RTA), hematuria, glycosuria, hypokalemia, and impaired urinary concentrating ability [3, 6, 17, 22]. Furthermore, nephrolithiasis, nephrocalcinosis, and various types of glomerulonephritis (GN) have been reported, often requiring immunosuppressive treatments [6, 23].

Pathophysiology
A genetic predisposition and aberrant activation of the immune system are responsible for the development of glandular and EGMs in SS [24]. In affected patients, there is an abnormal activation of CD4 + T cell-derived and NK cells, mostly caused by an altered interferon (IFN)-λ signaling pathway upon stimulation by pro-inflammatory cytokines such as IL-12, IL-23, and IL-33 [24]. Moreover, IL-17 and IL-22, predominantly secreted by Th17 cells, promote inflammation via IL-6 and tumor necrosis factor (TNF)-α and, indirectly, BAFF production. This results in MHC-II overexpression, increased lymphocytic infiltration, and, controversially, an overproduction of multiple autoantibodies, including anti-Ro/SSA and anti-La/SSB [24]. In addition, it has been proposed that certain genes involved in the Wnt/β-catenin signaling pathway may be involved in the pathogenesis of SS [24]. In patients with kidney involvement, the histologic findings are predominantly represented by an interstitial infiltration of T cells, B lymphocytes, and plasma cells [25]. Notably, a younger onset age in adults correlates with a higher prevalence of B-cell-mediated glandular and EGMs, including kidney involvement [25]. The high incidence of lymphopenia observed in a substantial number of patients is accompanied by lymphoplasmacytic infiltrates in the kidney interstitium [25]. In patients with kidney involvement, the association with lymphomas and the key role of IL36g and cryoglobulins may be crucial factors to understand in the future [25].
Clinical features
Pediatric SS is an extremely uncommon disorder that predominantly affects older children and adolescents, with a strong female predominance (80–87%), comparable to that observed in adult patients [7,8,9,10, 16]. In adults, SS is typically diagnosed between 30 and 50 years of age, whereas in children, the average age at diagnosis ranges from 12 to 14.2 years, and the primary form accounts for nearly all cases [2, 3, 7,8,9, 16, 18]. Pediatric SS is characterized by a variable phenotype whose characteristics and severity can vary markedly between patients [3, 13, 26]. The clinical findings of pediatric SS can be divided into glandular manifestations, which predominantly involve the parotid glands, and EGMs.
Glandular manifestations
The most common feature in childhood is recurrent parotid enlargement, which is present in up to 64% of patients, although submandibular gland swelling may also be observed [3, 7,8,9,10, 16, 20]. Bilateral or unilateral salivary gland enlargement is frequently accompanied by pain and tenderness and, unlike adults, has not been associated with increased mortality rate [3, 27]. Dryness and sicca syndrome are also common findings in children, with prevalence rates ranging from 34 to 80% [2, 3, 7,8,9,10, 15, 16, 18]. Patients with sicca syndrome experience dry eyes and mouth, resulting in an inability to assimilate food, halitosis, mucositis, weight loss, decreased tear production, and keratoconjunctivitis sicca up to corneal ulcerations [3, 18, 28]. Despite the paucity of distinct epidemiological data, it has been reported that children with SS can suffer from recurrent vaginitis, pancreatitis, chronic dry wheezing, and dry skin [3, 18, 29].
Extraglandular manifestations
A deeper understanding of EGMs of pediatric SS is currently a major area of focus for pediatric rheumatologists [3, 9, 18]. Musculoskeletal manifestations are the most prevalent EGMs, ranging from 21 to 58% of cases. Their clinical spectrum includes myalgias, arthralgias, and severe forms of arthritis and myositis [9]. Arthralgias are the most prevalent musculoskeletal involvement in children, affecting 54% of patients, whereas nonerosive arthritis affects large joints in 24% of cases [3, 9]. In addition, constitutional symptoms, such as fever, affect up to 41% of children with SS [3, 7,8,9,10, 16, 20]. Fatigue and malaise are frequent symptoms, with an unknown etiopathogenesis that is likely influenced by both chronic pain and depression [3, 18]. Pulmonary manifestations have been reported in less than 8% of pediatric patients, with less severe complications than in adults [3, 8, 9, 16]. The main respiratory finding is interstitial lung disease, mainly resulting in chronic cough and, on rare occasions, dyspnea [3]. In addition, skin manifestations have been reported in up to 51.3% of children with SS, including dryness, Raynaud’s phenomenon, cutaneous vasculitis, purpura, erythema nodosum, and cutaneous amyloidosis [3, 8, 18]. The gastrointestinal tract is primarily implicated in oral and pancreatic dysfunction. However, chronic diarrhea, nonalcoholic steatohepatitis, atrophic gastritis, and chronic gastritis have also been described in pediatric SS [3, 18, 30]. Neurologic complications have been reported in up to 17% of pediatric patients [8, 10, 16]. Interestingly, neuromyelitis optica spectrum disorder and SS have been recently investigated due to their unusual co-occurrence in childhood [3, 31,32,33]. Other central nervous system implications include encephalopathy and meningoencephalitis, resulting in headache, vomiting, chorea, paralysis, and seizures [3, 17, 34,35,36]. The peripheral nervous system can also be implicated, with polyneuropathy and mononeuritis [3, 18]. Hematologic complications affect 17–35.9% of children with SS, including mild chronic anemia, thrombocytopenia, and leukopenia, up to lymphomas [3, 18, 20, 37]. According to the literature, patients with pSS have a 10–44 fold greater risk of lymphoma than the general population, even higher than patients with SLE and RA [38]. Up to 10% of patients with pSS develop low-grade B cell lymphoma, typically extranodal marginal zone lymphomas (eMZL) of mucosa-associated lymphoid tissue (MALT) [38, 39]. Interestingly, the development of eMZL is often related to cryoglobulinemia and seems to be more frequent in adult pSS with kidney involvement than those without kidney manifestations [17, 40, 41]. There is controversy as to whether there is a higher incidence of lymphoma in patients with TIN or GN, with studies producing contradictory results [40, 41]. Kidney involvement in pediatric SS represents this paper’s main argument and is described in detail in the following sections.
Kidney involvement in children with Sjögren’s syndrome
The prevalence of kidney involvement in pediatric SS is estimated between 5 and 20.5%, and both tubulointerstitial and glomerular damage have been described [7,8,9,10, 16, 20, 21]. The clinical spectrum is wide and can range from latent to life-threatening conditions, mostly due to electrolyte disturbances. However, TIN is the most prevalent form of nephropathy in both adults and children with SS [6, 17, 19, 22]. According to a recent large multicenter study, TIN accounts for up to 98% of kidney manifestations in adults with SS who underwent a biopsy, similar to pediatric reports [6, 17, 18, 42]. In pediatric SS, a kidney biopsy should be considered when significant kidney involvement is suspected, such as in the presence of impaired urinary concentrating ability, evidence of RTA, hypokalemic paralysis, idiopathic recurrent nephrocalcinosis/nephrolithiasis, and proteinuria, particularly if intense (> 2 g/day or urinary protein/creatinine ratio > 2), suggesting a glomerular disease.
Tubulointerstitial involvement
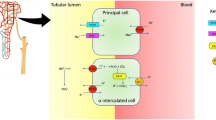
Chronic inflammation in TIN is caused by interstitial reactive lymphocytic infiltrates that are predominantly composed of B cells with sporadic T and NK lymphocytes, similar to the lymphocyte aggregates observed around the ducts of salivary and lacrimal glands, which are frequently organized into germinal center-like structures [6, 17, 19, 23]. When performed, kidney biopsy often reveals focal tubular atrophy, acinus damage, interstitial fibrosis, tubular epithelial cell degeneration, and fusion [17, 23]. In a recent review focusing on kidney involvement in pediatric SS, 10/21 patients underwent a kidney biopsy, and TIN was detected in 80% of cases [17]. TIN is often manifested by RTA, which is a well-known feature of kidney involvement in adult pSS, with estimated prevalence rates ranging from 62 to 73% [22, 43, 44]. Three major subtypes of RTA have been identified: proximal RTA (pRTA), which typically results from defective bicarbonate reabsorption in the proximal convoluted tubule; distal RTA (dRTA); and rare mixed forms [18, 22]. The specific pathophysiology underlying dRTA in SS has not yet been completely understood, and both the lack of H( +)-ATPase in the cortical collecting tubule, with a subsequent failure of the proton pump, and a permeability defect, resulting in bicarbonate leakage into the tubular lumen, have been suggested [6, 45, 46]. In addition, TIN and nephrocalcinosis may worsen medullary ammonia transfer [6]. In adult SS, dRTA occurs in over 95% of RTA cases, with a global prevalence in pSS patients ranging from 4 to 39%, predominantly in middle-aged women [6, 43, 47]. In particular, the alpha-intercalated cells of the collecting duct fail to secrete a sufficient amount of hydrogen ions in the urine, leading to a non-anion-gap metabolic acidosis with alkaline urine [18, 19]. Other cations such as potassium, calcium, and phosphorus are secreted to maintain electroneutrality, resulting in hypokalemia, nephrolithiasis, and bone resorption (Fig. 2) [18, 48, 49]. Thus, RTA is considered one of the main kidney manifestations of TIN in pediatric SS [6, 17]. In a cohort of 12 pediatric patients with RTA and SS, RTA was most frequently associated with pSS and present from the onset (in 25% of patients) to 9 years after diagnosis, affecting older children and adolescents [22]. Notably, dRTA is the most common subtype of RTA also in pediatric SS, while pRTA and mixed forms have rarely been observed [6, 17, 22]. However, the global prevalence of RTA in pSS is estimated to be approximately 9% [6]. Interestingly, only 14 cases of RTA had been detected in pediatric SS in a report from 2020, and the majority of them had dRTA [6, 17, 22, 34]. In this cohort, the associated kidney diseases were diabetes insipidus and nephrocalcinosis in four and three cases, respectively, with both occurring simultaneously in one patient [6, 17, 22]. The main manifestations of RTA in pediatric SS are non-anion gap metabolic acidosis, dehydration, and hypokalemia with alkaline urine [6, 17, 22, 50]. Hypokalemia is believed to occur in up to 92% of children with RTA and SS, with the lowest reported serum values of 1.2 mEq/L, whereas serum bicarbonate had reached reported values of 9.6 mg/dL [22]. Despite the fact that severe hypokalemia can occur in such children, also with a prolonged QT interval, an asymptomatic course can be observed [6]. The most feared complication of severe hypokalemia in pediatric SS is hypokalemic periodic paralysis, which causes profound weakness or paralysis; according to small case series, this life-threatening condition has been reported in up to half of children with RTA and SS, predominantly female adolescents and older children [6, 17, 22, 47, 51, 52]. To date, including adults, about 60 cases of hypokalemic paralysis in SS patients with RTA, mostly dRTA, have been reported [47, 51, 53,54,55]. Significantly, hypokalemic paralysis is regarded as the initial symptom in 7% of SS patients, according to a series of 130 individuals [43, 47].

Tubulointerstitial involvement. In tubulointerstitial involvement, kidney biopsy usually documents interstitial lymphocyte aggregates with multiple findings, such as focal tubular atrophy, acinus damage, interstitial fibrosis, tubular epithelial cell degeneration and fusion. Tubulointerstitial nephritis mainly presents with renal tubular acidosis with various degrees of polyuria and proteinuria. A failure of the proton pump, together with a permeability defect, are thought to be the cause of an impaired hydrogen secretion, resulting in non-anion-gap metabolic acidosis with alkaline urine. Thus, other cations such as potassium, calcium, and phosphorus are secreted to maintain electroneutrality. Hypokalemia can be mild or severe, leading to ECG abnormalities, weakness, up to hypokalemic periodic paralysis. The large amount of calcium and phosphorus secreted can be responsible for bone resorption, nephrolithiasis and nephrocalcinosis
Although several cases have been reported in adults, nephrocalcinosis is a very rare finding in SS children, and only two cases have been reported to date [6, 56]. Furthermore, nephrogenic diabetes insipidus has been described in pediatric SS, and affected patients can suffer from impaired urine-concentrating ability of varying degrees, including mild forms [6].
Glomerular damage
Glomerular involvement in children with SS is extremely rare and frequently associated with immunosuppressive treatments [6, 17]. To the best of our knowledge, only eight cases of predominant glomerular damage have been reported in pediatric SS in the literature, excluding forms of lupus nephritis in secondary SS associated with juvenile SLE (Table 2). However, glomerular involvement is an uncommon finding in adults with SS [18]. In a large series of 95 adult patients with SS and kidney involvement who underwent a kidney biopsy, 23.2% were found to have glomerular damage, frequently in conjunction with TIN [57]. Multiple bioptic findings, including minimal change disease, focal segmental and global glomerulosclerosis, crescentic GN, membranous nephropathy, membranoproliferative glomerulonephritis, and IgA nephropathy, have been documented in such patients [17, 19, 40, 43, 58, 59].
Diagnostic algorithm
The absence of specific classification criteria for pediatric forms is a major issue [6, 60, 61]. Indeed, there is growing consensus that EGMs and systemic involvement are more common in pediatric SS than in the adult form. In addition, the early manifestations of pediatric SS are heterogenous, making early diagnosis challenging, resulting in underdiagnosis and prolonged diagnostic delays [6, 20, 21, 60]. The historical American–European Consensus Group (AECG) criteria for pSS were developed and validated between 1989 and 1996, and improved in 2002 [62]. The last revision of these classification criteria for pSS was validated in 2016 by the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR), and represents the current diagnostic tool for the disease [63]. According to the 2016 ACR/EULAR consensus, (1) anti-Ro/SSA antibody positivity, (2) focal lymphocytic sialadenitis, pathogenic ocular findings measured with (3) the Ocular Staining Score and (4) the Schirmer’s test, and (5) a lack of salivary flow rate represent the current classification criteria for SS [63]. In 1999, however, specific classification criteria for pediatric SS were proposed, with improvements on the diagnostic rate (in a series of 135 cases of pediatric SS, the diagnostic rate increased from 39 to 76% regarding the AECG criteria) [20, 60, 61]. Nonetheless, some characteristics are shared with the adult form of SS, such as reactive lymphocytic infiltration of the exocrine glands, anti-Ro/SSA and/or anti-La/SSB antibody positivity, antinuclear antibodies (ANA) positivity, polyclonal hypergammaglobulinemia, and high erythrocyte sedimentation rate (ESR) [6, 20]. According to a systematic review of the literature on pediatric pSS, ANA (63.6–96.2%), anti-Ro/SSA (36.4–84.6%), and anti-La/SSB (27.3–65.4%) are highly prevalent in children with SS [64]. In addition, hyperglobulinemia is a common finding in children with SS as a result of heightened B lymphocyte activation [17, 65]. Kidney involvement is not included in the 2016 ACR/EULAR criteria for SS classification but, as discussed above, kidney manifestations play an important role in pediatric SS, and manifest or latent dRTA had been included in the classification criteria proposed for pediatric pSS in 1999 [60]. In clinical practice, hematuria, proteinuria, and alkaline urine may suggest tubulointerstitial or glomerular damage in children. Specifically, tubulointerstitial inflammation can be suggested by an increased urinary excretion of β-2 microglobulin (β2M) at the diagnosis and during follow-up and its normalization can reflect improvement after therapy [6]. Blood exams should routinely be performed to detect increased ESR, non-anion gap metabolic acidosis, hypokalemia, and other electrolyte disfunctions, serum creatinine, serum proteins, and albumin. Kidney ultrasonography may be performed to investigate nephrocalcinosis, nephrolithiasis, and other morphological abnormalities. Lastly, a kidney biopsy may be useful for determining the nature and extent of kidney damage and the need for a specific treatment (Fig. 3) [6, 17].

Flow-chart illustrating a proposed diagnostic approach for pediatric Sjögren’s syndrome, with a focus on kidney involvement. RTA, renal tubular acidosis
Treatment options and outcome
Despite the lack of strong recommendations due to the rarity of the disease, adult patients with SS are treated with immunosuppressive drugs and nonpharmacologic measures [24, 66]. Artificial tears and punctal plugs may be beneficial for xerophthalmia, whereas intra-oral lubricating devices do not appear to be effective for xerostomia [66]. Low-dose glucocorticoids (GCs) may be helpful in moderate systemic activity forms, despite the frequent occurrence of adverse effects in long-term treatments [67]. Adult patients with SS are commonly treated with conventional disease-modifying antirheumatic drugs (DMARDs), such as hydroxychloroquine (HCQ; mainly for its efficacy in SLE), methotrexate (MTX; especially for musculoskeletal involvement), azathioprine (AZA), cyclosporine (CSA), and mycophenolate mofetil (MMF); however, the improvements are slight and controversial [68, 69]. In some cases, monoclonal antibodies may also be beneficial for adult SS, particularly rituximab, even though a reduction in fatigue and dehydration has not been conclusively demonstrated [70]. Inhibitors of IL-1, IL-6, and TNF-α did not show significant improvements; nevertheless, several ongoing studies are evaluating potential future treatment strategies [24].
According to a systematic review of the literature, multiple treatment strategies for children with SS have been reported, all of which are based on scant evidence of efficacy from small series and case reports. Thus, there is a lack of good-quality studies and guidelines for a correct, evidence-based approach to pediatric SS, probably also due to the widely variable phenotype [26]. Such concerns may also be found in the current adult guidelines for the management of pSS, released by the British Society for Rheumatology in 2017 [26, 71]. GCs, DMARDs, including HCQ and MTX, and nonsteroidal anti-inflammatory drugs are the most common drugs used in patients with pSS [26]. Kidney involvement shares the same treatment limitations as other manifestations of the disease. GCs, mostly prednisone and methylprednisolone, are widely used in both tubulointerstitial and glomerular damage in children with SS [6, 17, 22, 26, 34, 50, 56, 72, 73]. Significantly, GCs are administered in about 52% of pediatric SS patients with EGMs, and one patient with hypokalemic paralysis due to dRTA required high doses of methylprednisolone (100 mg/day) [26]. Good improvement has been noted after the administration of oral prednisone in a consistent number of patients, but its efficacy alone is a controversial issue due to the frequent co-administration of other DMARDs [26]. According to the literature, 62% of children with SS have been treated with DMARDs, including HCQ (34%), MTX (5.8%), and others [26]. Kidney implications require a prompt treatment strategy. Stable kidney function has been detected after a multi-drug therapy with GCs, MMF, and AZA [6]. A combination of GCs and cyclophosphamide has also been reported in a few patients, with various outcomes [17, 46, 56, 74]. One patient with dRTA was successfully treated with GCs and CSA [52]. Kidney damage in children with SS has also been treated with HCQ, MTX, and rituximab, often with good improvement [17, 32, 50]. In order to correct acidosis and hypokalemia, alkali and electrolyte supplementation (such as potassium chloride or citrate) is usually required [6, 26]. Despite clinical improvement being reported following treatment with various drugs, this has not been studied in a controlled setting and may not be generalizable to all children with SS; therefore, no conclusions can be made regarding the efficacy of therapy. However, although the long-term prognosis of kidney involvement in pediatric pSS is usually favorable, chronic immunosuppression is often required, and even kidney failure has been described suggesting that SS may represent an underdiagnosed cause of kidney failure in children [17, 23].
Conclusions
EGMs are a common issue in children with SS, and among these, kidney involvement plays a relevant role. Kidney implications may include both tubulointerstitial and glomerular damage, and a biopsy may reveal multiple different findings. Pediatric SS should be suspected in older children and adolescents, especially females, with hematuria, proteinuria, or impaired urinary concentrating ability, associated with salivary gland swelling and positive rates of ANA and anti-Ro/SSA antibodies. In addition, rare but life-threatening conditions may be observed, including periodic hypokalemic paralysis. Although the prognosis of kidney involvement in pediatric SS is typically favorable, the risk of developing lymphomas seems to be greater, prolonged immunosuppressive therapies are usually necessary, and kidney failure can occur. Thus, it is critical to improve our knowledge about kidney involvement in pediatric SS to provide a prompt diagnosis and improve the disease management.
Key summary points
-
1.
Pediatric Sjögren’s syndrome accounts for approximately 1% of all forms of SS, representing a rare autoimmune disease in childhood.
-
2.
The main clinical feature in children with Sjögren’s syndrome is salivary gland enlargement, although sicca syndrome is also commonly observed. On the other hand, extraglandular manifestations are more significant than in adults.
-
3.
Tubulointerstitial nephritis is the most frequent kidney injury in affected children, leading to kidney tubular acidosis and electrolyte disturbances such as severe hypokalemia, which can lead to periodic hypokalemic paralysis.
-
4.
Glomerular damage is less common than tubular interstitial involvement. Several types of glomerulonephritis have been observed at kidney biopsy, and immunosuppressive drugs are usually required.
-
5.
Treatment strategies for the kidney implications of pediatric Sjögren’s syndrome lack strong recommendations and are based on case reports and case series. Specific classification criteria for children are needed to increase the diagnostic rate and improve outcomes.
Multiple choice questions
Answers are given following the reference list.
-
1.
Which of the following statements is true about pediatric Sjögren’s syndrome?
-
A)
Sicca syndrome is the most significant feature.
-
B)
Kidney involvement is not relevant in affected patients.
-
C)
Kidney failure has been observed due to severe kidney involvement.
-
D)
Current classification criteria are accurate enough for the pediatric population.
-
A)
-
2.
What percentage of children with Sjögren’s syndrome has kidney manifestations?
-
A)
5–20.5%
-
B)
1–5%
-
C)
45–70%
-
D)
0.5–2%
-
A)
-
3.
Which of the following manifestations of tubulointerstitial nephritis is the most feared in children?
-
A)
Macroscopic hematuria
-
B)
Mild impaired urinary concentrating ability
-
C)
Periodic hypokalemic paralysis
-
D)
Severe proteinuria
-
A)
-
4.
What is the most serious long-term complication in pediatric Sjögren’s syndrome with kidney involvement?
-
A)
Recurrent nephrolithiasis
-
B)
Persistent proteinuria
-
C)
Diabetes insipidus
-
D)
Extranodal marginal zone lymphomas
-
A)
-
5.
Which of the following classification criteria for Sjögren’s syndrome includes kidney manifestation?
-
A)
The 2016 ACR/EULAR criteria for all patients with Sjögren’s syndrome
-
B)
The 1999 criteria for children and adolescents with Sjögren’s syndrome
-
C)
The 2002 AECG criteria for all patients with Sjögren’s syndrome
-
D)
None of these
-
A)
Data availability
Not applicable to this article as no datasets were generated or analyzed during the current study.
References
Negrini S, Emmi G, Greco M et al (2022) Sjögren’s syndrome: a systemic autoimmune disease. Clin Exp Med 22:9–25. https://doi.org/10.1007/s10238-021-00728-6
Brito-Zerón P, Baldini C, Bootsma H et al (2016) Sjögren syndrome. Nat Rev Dis Primer 2:16047. https://doi.org/10.1038/nrdp.2016.47
Randell RL, Lieberman SM (2021) Unique aspects of pediatric Sjögren disease. Rheum Dis Clin North Am 47:707–723. https://doi.org/10.1016/j.rdc.2021.07.008
Zhong H, Liu S, Wang Y et al (2022) Primary Sjögren’s syndrome is associated with increased risk of malignancies besides lymphoma: a systematic review and meta-analysis. Autoimmun Rev 21:103084. https://doi.org/10.1016/j.autrev.2022.103084
Fox RI (2005) Sjögren’s syndrome. Lancet 366:321–331. https://doi.org/10.1016/S0140-6736(05)66990-5
Bogdanović R, Basta-Jovanović G, Putnik J et al (2013) Renal involvement in primary Sjogren syndrome of childhood: case report and literature review. Mod Rheumatol 23:182–189. https://doi.org/10.1007/s10165-012-0633-x
Means C, Aldape MA, King E (2017) Pediatric primary Sjögren syndrome presenting with bilateral ranulas: a case report and systematic review of the literature. Int J Pediatr Otorhinolaryngol 101:11–19. https://doi.org/10.1016/j.ijporl.2017.07.019
Hammenfors DS, Valim V, Bica BERG et al (2020) Juvenile Sjögren’s syndrome: clinical characteristics with focus on salivary gland ultrasonography. Arthritis Care Res 72:78–87. https://doi.org/10.1002/acr.23839
Basiaga ML, Stern SM, Mehta JJ et al (2021) Childhood Sjögren syndrome: features of an international cohort and application of the 2016 ACR/EULAR classification criteria. Rheumatology (Oxford) 60:3144–3155. https://doi.org/10.1093/rheumatology/keaa757
Marino A, Romano M, Giani T et al (2021) Childhood Sjogren’s syndrome: an Italian case series and a literature review-based cohort. Semin Arthritis Rheum 51:903–910. https://doi.org/10.1016/j.semarthrit.2020.11.004
Nikitakis NG, Rivera H, Lariccia C et al (2003) Primary Sjögren syndrome in childhood: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 96:42–47. https://doi.org/10.1016/s1079-2104(03)00159-8
Civilibal M, Canpolat N, Yurt A et al (2007) A child with primary Sjögren syndrome and a review of the literature. Clin Pediatr (Phila) 46:738–742. https://doi.org/10.1177/0009922807301945
Singer NG, Tomanova-Soltys I, Lowe R (2008) Sjögren’s syndrome in childhood. Curr Rheumatol Rep 10:147–155. https://doi.org/10.1007/s11926-008-0026-5
Schuetz C, Prieur A-M, Quartier P (2010) Sicca syndrome and salivary gland infiltration in children with autoimmune disorders: when can we diagnose Sjögren syndrome? Clin Exp Rheumatol 28:434–439
Cimaz R, Casadei A, Rose C et al (2003) Primary Sjögren syndrome in the paediatric age: a multicentre survey. Eur J Pediatr 162:661–665. https://doi.org/10.1007/s00431-003-1277-9
Ramos-Casals M, Acar-Denizli N, Vissink A et al (2021) Childhood-onset of primary Sjögren’s syndrome: phenotypic characterization at diagnosis of 158 children. Rheumatology (Oxford) 60:4558–4567. https://doi.org/10.1093/rheumatology/keab032
Zhao J, Chen Q, Zhu Y et al (2020) Nephrological disorders and neurological involvement in pediatric primary Sjogren syndrome: a case report and review of literature. Pediatr Rheumatol Online J 18:39. https://doi.org/10.1186/s12969-020-00431-y
Vivino FB, Bunya VY, Massaro-Giordano G et al (2019) Sjogren’s syndrome: an update on disease pathogenesis, clinical manifestations and treatment. Clin Immunol 203:81–121. https://doi.org/10.1016/j.clim.2019.04.009
François H, Mariette X (2016) Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol 12:82–93. https://doi.org/10.1038/nrneph.2015.174
Gong Y, Liu H, Li G et al (2023) Childhood-onset primary Sjögren’s syndrome in a tertiary center in China: clinical features and outcome. Pediatr Rheumatol Online J 21:11. https://doi.org/10.1186/s12969-022-00779-3
Liu C, Jin Y, Huang H et al (2022) Clinical and laboratory features of childhood-onset primary Sjögren’s syndrome: a retrospective study from China. Front Pediatr 10:1044812. https://doi.org/10.3389/fped.2022.1044812
Pessler F, Emery H, Dai L et al (2006) The spectrum of renal tubular acidosis in paediatric Sjögren syndrome. Rheumatology (Oxford) 45:85–91. https://doi.org/10.1093/rheumatology/kei110
Johnson S, Hulton S-A, Brundler M-A et al (2007) End-stage renal failure in adolescence with Sjögren’s syndrome autoantibodies SSA and SSB. Pediatr Nephrol 22:1793–1797. https://doi.org/10.1007/s00467-007-0526-y
Zhan Q, Zhang J, Lin Y et al (2023) Pathogenesis and treatment of Sjögren’s syndrome: review and update. Front Immunol 14:1127417. https://doi.org/10.3389/fimmu.2023.1127417
Chatterjee R, Balakrishnan A, Kharbanda R et al (2023) Renal involvement in Sjögren’s syndrome: predictors and impact on patient outcomes. Rheumatol Int 43:1297–1306. https://doi.org/10.1007/s00296-022-05242-w
Doolan G, Faizal NM, Foley C et al (2022) Treatment strategies for Sjögren’s syndrome with childhood onset: a systematic review of the literature. Rheumatology (Oxford) 61:892–912. https://doi.org/10.1093/rheumatology/keab579
Singh AG, Singh S, Matteson EL (2016) Rate, risk factors and causes of mortality in patients with Sjögren’s syndrome: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford) 55:450–460. https://doi.org/10.1093/rheumatology/kev354
Akpek EK, Mathews P, Hahn S et al (2015) Ocular and systemic morbidity in a longitudinal cohort of Sjögren’s syndrome. Ophthalmology 122:56–61. https://doi.org/10.1016/j.ophtha.2014.07.026
van Nimwegen JF, van der Tuuk K, Liefers SC et al (2020) Vaginal dryness in primary Sjögren’s syndrome: a histopathological case-control study. Rheumatology (Oxford) 59:2806–2815. https://doi.org/10.1093/rheumatology/keaa017
Kashiwagi Y, Hatsushika T, Tsutsumi N et al (2017) Gastrointestinal and liver lesions in primary childhood Sjögren syndrome. Clin Rheumatol 36:1433–1435. https://doi.org/10.1007/s10067-017-3599-4
Gmuca S, Lieberman SM, Mehta J (2017) Pediatric neuromyelitis optica spectrum disorder and Sjögren syndrome: more common than previously thought? J Rheumatol 44:959–960. https://doi.org/10.3899/jrheum.160978
Kornitzer JM, Kimura Y, Janow GL (2016) Primary Sjögren syndrome in a child with a neuromyelitis optica spectrum disorder. J Rheumatol 43:1260–1261. https://doi.org/10.3899/jrheum.151207
Fang G-L, Fang W, Zheng Y, Zhang Y-X (2021) Neuromyelitis optica spectrum disorder complicated with Sjögren’s syndrome: first pediatric case responsive to mycophenolate mofetil treatment. Acta Neurol Belg 121:1065–1067. https://doi.org/10.1007/s13760-020-01530-z
Matsui Y, Takenouchi T, Narabayashi A et al (2016) Childhood Sjögren syndrome presenting as acute brainstem encephalitis. Brain Dev 38:158–162. https://doi.org/10.1016/j.braindev.2015.05.005
Iwai K, Amo K, Kuki I et al (2019) An unusual manifestation of Sjögren syndrome encephalitis. Brain Dev 41:217–220. https://doi.org/10.1016/j.braindev.2018.08.004
Delorme C, Cohen F, Hubsch C, Roze E (2015) Chorea as the initial manifestation of Sjögren syndrome. Pediatr Neurol 52:647–648. https://doi.org/10.1016/j.pediatrneurol.2015.02.021
Nakahara E, Yagasaki H, Shimozawa K et al (2016) Severe thrombocytopenia as initial signs of primary Sjögren syndrome in a 9-year-old female. Pediatr Blood Cancer 63:1312–1313. https://doi.org/10.1002/pbc.25977
Retamozo S, Brito-Zerón P, Ramos-Casals M (2019) Prognostic markers of lymphoma development in primary Sjögren syndrome. Lupus 28:923–936. https://doi.org/10.1177/0961203319857132
Kolijn PM, Huijser E, Wahadat MJ et al (2023) Extranodal marginal zone lymphoma clonotypes are detectable prior to eMZL diagnosis in tissue biopsies and peripheral blood of Sjögren’s syndrome patients through immunogenetics. Front Oncol 13:1130686. https://doi.org/10.3389/fonc.2023.1130686
Goules AV, Tatouli IP, Moutsopoulos HM, Tzioufas AG (2013) Clinically significant renal involvement in primary Sjögren’s syndrome: clinical presentation and outcome. Arthritis Rheum 65:2945–2953. https://doi.org/10.1002/art.38100
Narvaez J, Sánchez-Piedra C, Fernández-Castro M et al (2020) Clinically significant renal involvement in primary Sjögren’s syndrome is associated with important morbidity: data from the Spanish Sjögrenser cohort. Clin Exp Rheumatol 38(Suppl 126):116–124
Jasiek M, Karras A, Le Guern V et al (2017) A multicentre study of 95 biopsy-proven cases of renal disease in primary Sjögren’s syndrome. Rheumatology (Oxford) 56:362–370. https://doi.org/10.1093/rheumatology/kew376
Ren H, Wang W-M, Chen X-N et al (2008) Renal involvement and followup of 130 patients with primary Sjögren’s syndrome. J Rheumatol 35:278–284
Kaufman I, Schwartz D, Caspi D, Paran D (2008) Sjögren’s syndrome - not just Sicca: renal involvement in Sjögren’s syndrome. Scand J Rheumatol 37:213–218. https://doi.org/10.1080/03009740701867323
Cohen EP, Bastani B, Cohen MR et al (1992) Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol 3:264–271. https://doi.org/10.1681/ASN.V32264
Zawadzki J (1998) Permeability defect with bicarbonate leak as a mechanism of immune-related distal renal tubular acidosis. Am J Kidney Dis 31:527–532. https://doi.org/10.1053/ajkd.1998.v31.pm9506692
Garza-Alpirez A, Arana-Guajardo AC, Esquivel-Valerio JA et al (2017) Hypokalemic paralysis due to primary Sjögren syndrome: case report and review of the literature. Case Rep Rheumatol 2017:7509238. https://doi.org/10.1155/2017/7509238
Evans R, Zdebik A, Ciurtin C, Walsh SB (2015) Renal involvement in primary Sjögren’s syndrome. Rheumatol Oxf Engl 54:1541–1548. https://doi.org/10.1093/rheumatology/kev223
Jain A, Srinivas BH, Emmanuel D et al (2018) Renal involvement in primary Sjögren’s syndrome: a prospective cohort study. Rheumatol Int 38:2251–2262. https://doi.org/10.1007/s00296-018-4118-x
Ohlsson V, Strike H, James-Ellison M et al (2006) Renal tubular acidosis, arthritis and autoantibodies: primary Sjögren’s syndrome in childhood. Rheumatology (Oxford) 45:238–240. https://doi.org/10.1093/rheumatology/kei175
Bruns N, Finkelberg I, Al-Attrach I et al (2020) Unusual presentation of polyautoimmunity and renal tubular acidosis in an adolescent with Hashimoto’s thyroiditis and central pontine myelinolysis. Front Endocrinol 11:548877. https://doi.org/10.3389/fendo.2020.548877
Skalova S, Minxova L, Slezak R (2008) Hypokalaemic paralysis revealing Sjögren’s syndrome in a 16-Year Old Girl. Ghana Med J 42:124–128
Fazal F, Ur Rehman ME, Tahir S et al (2022) Hypokalemic quadriparesis as initial presentation of secondary Sjogren syndrome with associated autoimmune thyroiditis: a case report. Cureus 14:e25420. https://doi.org/10.7759/cureus.25420
Permatasari CA, Zahraini H, Marpaung FR, Aryati (2022) Hypokalemic periodic paralysis and renal tubular acidosis in a patient with hypothyroid and autoimmune disease. Ann Med Surg (Lond) 75:103389. https://doi.org/10.1016/j.amsu.2022.103389
Meena DS, Kumar D, Bohra GK, Bhambu SK (2020) Hypokalemic paralysis as an initial presentation of Sjogren syndrome. Ann Afr Med 19:147–149. https://doi.org/10.4103/aam.aam_34_19
Kobayashi I, Furuta H, Tame A et al (1996) Complications of childhood Sjögren syndrome. Eur J Pediatr 155:890–894. https://doi.org/10.1007/BF02282840
Jung SK, Park KH, Yim HE et al (2010) Primary Sjögren’s syndrome with mesangial proliferative glomerulonephritis and IgA deposits in a child. Pediatr Nephrol 25:567–568. https://doi.org/10.1007/s00467-009-1358-8
Kidder D, Rutherford E, Kipgen D et al (2015) Kidney biopsy findings in primary Sjögren syndrome. Nephrol Dial Transplant 30:1363–1369. https://doi.org/10.1093/ndt/gfv042
Lin D-F, Yan S-M, Zhao Y et al (2010) Clinical and prognostic characteristics of 573 cases of primary Sjögren’s syndrome. Chin Med J (Engl) 123:3252–3257
Bartůnková J, Sedivá A, Vencovský J, Tesar V (1999) Primary Sjögren’s syndrome in children and adolescents: proposal for diagnostic criteria. Clin Exp Rheumatol 17:381–386
Houghton K, Malleson P, Cabral D et al (2005) Primary Sjögren’s syndrome in children and adolescents: are proposed diagnostic criteria applicable? J Rheumatol 32:2225–2232
Vitali C, Bombardieri S, Jonsson R et al (2002) Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 61:554–558. https://doi.org/10.1136/ard.61.6.554
Shiboski CH, Shiboski SC, Seror R et al (2017) 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis 76:9–16. https://doi.org/10.1136/annrheumdis-2016-210571
Virdee S, Greenan-Barrett J, Ciurtin C (2017) A systematic review of primary Sjögren’s syndrome in male and paediatric populations. Clin Rheumatol 36:2225–2236. https://doi.org/10.1007/s10067-017-3745-z
Kang KY, Kim H-O, Kwok S-K et al (2011) Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res Ther 13:R179. https://doi.org/10.1186/ar3504
Hackett KL, Deane KHO, Strassheim V et al (2015) A systematic review of non-pharmacological interventions for primary Sjögren’s syndrome. Rheumatology (Oxford) 54:2025–2032. https://doi.org/10.1093/rheumatology/kev227
Gheitasi H, Kostov B, Solans R et al (2015) How are we treating our systemic patients with primary Sjögren syndrome? Analysis of 1120 patients. Int Immunopharmacol 27:194–199. https://doi.org/10.1016/j.intimp.2015.03.027
Fauchais A-L, Ouattara B, Gondran G et al (2010) Articular manifestations in primary Sjögren’s syndrome: clinical significance and prognosis of 188 patients. Rheumatology (Oxford) 49:1164–1172. https://doi.org/10.1093/rheumatology/keq047
Price EJ, Rigby SP, Clancy U, Venables PJ (1998) A double blind placebo controlled trial of azathioprine in the treatment of primary Sjögren’s syndrome. J Rheumatol 25:896–899
Wang X, Lin X, Su Y, Wang H (2023) Systematic review with meta-analysis: efficacy and safety of biological treatment on salivary gland function in primary Sjögren’s syndrome. Front Pharmacol 14:1093924. https://doi.org/10.3389/fphar.2023.1093924
Price EJ, Rauz S, Tappuni AR et al (2017) The British Society for Rheumatology guideline for the management of adults with primary Sjögren’s syndrome. Rheumatology (Oxford) 56:1828. https://doi.org/10.1093/rheumatology/kex375
Yoshida K, Suzuki J, Kume K et al (1996) Sjögren’s syndrome with membranous glomerulonephritis detected by urine screening of schoolchildren. Acta Paediatr Jpn 38:533–536. https://doi.org/10.1111/j.1442-200x.1996.tb03540.x
Kagan M, Bervina N, Vorobyeva O et al (2011) Pauci-immune crescentic glomerulonephritis complicating Sjögren’s syndrome in a 12-year-old girl. Pediatr Nephrol 26:991–992. https://doi.org/10.1007/s00467-011-1760-x
Berman JL, Kashii S, Trachtman MS, Burde RM (1990) Optic neuropathy and central nervous system disease secondary to Sjögren’s syndrome in a child. Ophthalmology 97:1606–1609. https://doi.org/10.1016/s0161-6420(90)32369-2
Ito S, Kamei K, Ikoma M (2010) Primary Sjögren syndrome that developed after IgA nephropathy. Pediatr Nephrol 25:1579–1580. https://doi.org/10.1007/s00467-010-1489-y
Author information
Authors and Affiliations
Contributions
SLB, ADL, and GDD: wrote the manuscript, realized the figures. LB, FC, and MV: coordinated, revised, and approved the final version of the manuscript. The content has not been published or submitted for publication elsewhere.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Answers: 1. C; 2. A; 3. C; 4. D; 5. B
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
La Bella, S., Vivarelli, M., Di Ludovico, A. et al. Kidney manifestations of pediatric Sjögren’s syndrome. Pediatr Nephrol 39, 711–721 (2024). https://doi.org/10.1007/s00467-023-06135-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-023-06135-1