
Abstract
Glomerular hyperfiltration (GHF) is a phenomenon that can occur in various clinical conditions affecting the kidneys such as sickle cell disease, diabetes mellitus, autosomal dominant polycystic kidney disease, and solitary functioning kidney. Yet, the pathophysiological mechanisms vary from one disease to another and are not well understood. More so, it has been demonstrated that GHF may occur at the single-nephron in some clinical conditions while in others at the whole-kidney level. In this review, we explore the pathophysiological mechanisms of GHF in relation to various clinical conditions in the pediatric population. In addition, we discuss the role and mechanism of action of important factors such as gender, low birth weight, and race in the pathogenesis of GHF. Finally, in this current review, we further highlight the consequences of GHF in the progression of kidney disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glomerular hyperfiltration (GHF) is generally defined as a supraphysiologic increase of glomerular filtration rate (GFR), although a precise definition is lacking [1]. GHF is associated with progressive loss of kidney function and is considered as a significant risk factor for developing chronic kidney disease (CKD) [2, 3]. It is also associated with adverse cardiovascular outcomes and all-cause mortality [4, 5]. Despite this association, the clinical implications and long-term consequences of GHF remain poorly evaluated, especially in the pediatric population [4].
GHF can result from an increase in factors determining GFR, such as kidney plasma flow, hydraulic pressure across the glomerular filtration barrier, or ultrafiltration coefficient [6, 7]. In physiologic states, it can occur after consumption of high protein meals or during pregnancy [8]. However, in pathologic states, GHF can be due to various diseases, either congenital or acquired. The pathophysiologic mechanisms leading to GHF depend on the underlying disease and are incompletely understood [8]. The present review discusses the different medical conditions causing GHF, their potential mechanisms, as well as the factors associated with the development of GHF, with a particular focus on the pediatric population. This review further highlights the potential and consequential damaging effect of GHF.
Prevalence of GHF in children
GHF is regarded as one of the early markers of kidney dysfunction for some forms of CKD, and it is used to predict progressive kidney-function decline. The exact prevalence of GHF in the general pediatric population remains relatively unknown, and this might be due to the paucity of studies determining GHF in healthy children as well as the lack of consensus on the GHF definition. The exact threshold for defining GHF is not well established in pediatric population, and it ranges from 120–180 ml/min per 1.73 m2 [9]. Regardless of these constraints, few pediatric studies have determined the prevalence of GHF in the general population. In a study to determine the metabolic risk factors in nondiabetic adolescents with GHF, Lee et al. reported that 11.8% of their study subjects had GHF when using 120 ml/min per 1.73 m2 as the threshold. The authors further showed that GHF is associated with hypertriglyceridemia and increased insulin resistance in their cohort [10]. It is worth mentioning that in this study, GFR was estimated by using the bedside Schwartz equation, which was designed for children with CKD, and therefore may be a less adequate formula for children without any medical condition [11, 12]. Nevertheless, another study using the full age spectrum (FAS) equation and 120 ml/min per 1.73 m2 as the threshold reported a similar prevalence (11.0%) of GHF in school-age children [13].
The pathophysiological mechanisms of GHF in relation to clinical conditions
In children, several clinical conditions are associated with increased GFR. These conditions can be classified into four groups based on the cause, namely: genetic diseases (e.g., sickle cell disease, autosomal dominant polycystic kidney disease, and Duchenne muscular dystrophy), metabolic disorders (such as diabetes mellitus), malnutrition (i.e., obesity), and solitary functioning kidney. The pathophysiological mechanisms mediating GHF differ from one disease to another. Nevertheless, it has been demonstrated that in some clinical conditions, GHF may occur at the single-nephron while in others at the whole-kidney level as illustrated in Fig. 1 [8, 14]. Regardless, it has been postulated that GHF occurs as a result of chronic arteriole vasodilation in the kidney [1], compensatory adaptation due to nephron loss, or the failure of tubuloglomerular feedback (TGF) [15].

Mechanisms of glomerular hyperfiltration. The pathophysiological mechanisms mediating glomerular hyperfiltration (GHF) vary from one clinical disease to another. In these diseases, GHF can either occur at the single-nephron level or at the whole-kidney level. GHF, glomerular hyperfiltration; KBF, kidney blood flow; FF, filtration fraction; NO, nitric oxide; HO-CO, heme oxygenase 1-carbon monoxide; RAAS, renin–angiotensin–aldosterone system; TGF, tubuloglomerular feedback
Sickle cell disease (SCD)
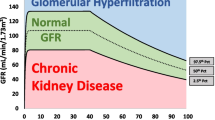
This is a genetic blood disorder caused by a point mutation in the β-globin gene, and it affects approximately 1 in 300,000 newborns annually [16, 17]. This point mutation leads to vaso-occlusion and hemolytic anemia, which are the characteristic hallmarks of the disease [18]. These hallmarks have been associated with the various multi-organ dysfunctions, including the sickle cell nephropathy [19]. Sickle cell nephropathy is the term given to all the kidney abnormalities associated with SCD. These abnormalities can be grouped into three main categories, isolated hematuria, tubular dysfunction, and glomerulopathy, and if left untreated can subsequently lead to CKD and kidney failure [18]. Glomerulopathy in children with SCD is well-described [18], with the earliest childhood clinical manifestation being GHF as demonstrated in infants (mean age = 13.7 months) recruited to the Pediatric Hydroxyurea Phase III Clinical Trial (BABY HUG) [20]. Indeed, the evolution of GFR in a population with SCD involves an initial increased GFR during early childhood, which then peaks at adolescence, before it finally begins to decline (Fig. 2a) [21, 22]. Though there is no well-established cut-off definition for GHF in the pediatric population and several methods have been used to measure or estimate GFR in SCD, the published data report the prevalence of GHF in pediatric populations ranging between 16% and 98%, as shown in Table 1 [9, 21, 23,24,25,26,27,28,29,30,31,32,33,34].

Glomerular hyperfiltration in sickle cell disease. a The evolution of glomerular filtration rate (GFR) in sickle cell disease (SCD) involves an increased GFR which begins during early childhood and peaks at adolescence. This increased GFR is usually followed by an elevated albumin excretion and subsequent decline in GFR in adulthood due to loss of kidney function. b The acidic, hypoxic, and hypertonic characteristic feature of the renal medulla promotes the sickling and polymerization of red blood cells (RBCs), resulting in vaso-occlusion, ischemia, and microinfarction of the vasa recta. This destruction leads to the release of vasodilating mediators (such as prostaglandins and nitric oxide, as well as the induction of the heme oxygenase 1-carbon monoxide (HO–CO) system) which instigate an increased vasodilation of the afferent arteriole (AA). This increased vasodilation of AA leads to an increased kidney blood flow (KPF), which in turn results in an increased glomerular capillary ultrafiltration coefficient (Kf), thus eventually leading to glomerular hyperfiltration. GFR, glomerular filtration rate; KBF, kidney blood flow; AA, afferent arteriole; EA, efferent arteriole; Kf, glomerular capillary ultrafiltration co-efficient; RBCs, red blood cells
In SCD, GHF has been associated with an increased kidney blood flow, a reduced filtration fraction, and an increased glomerular ultrafiltration coefficient [35,36,37]. This indicates that GHF is driven by dilatation of both afferent and efferent arterioles (with a predominant effect on the efferent arterioles due to a substantially greater increase in effective kidney blood flow compared to GFR) [38,39,40], increased glomerular perfusion, and glomerular enlargement (increased effective glomerular filtration surface area) [19]. To support this, the presence of abnormally distended enlarged glomeruli can be observed at the time of hyperfiltration in children with SCD as early as 7–18 months of age [41,42,43]. Furthermore, the rise in GFR has been suggested to be driven by increased cardiac output as a result of chronic anemia or by the localized increased release of kidney vasodilating prostaglandins (especially the high prostaglandin E2/F2α ratio, which may be physiologically relevant in regard to renin release [38]) and an increase in the nitric oxide (NO) synthase in response to the hypoxic condition in the renal medulla of individuals with SCD (Fig. 2b) [18, 19, 44, 45]. Although various findings support that chronic hemolysis confers risk for GHF [45, 46], the exact pathophysiological mechanisms remain unknown. Nevertheless, Nath et al. proposed that the chronic hemolysis in SCD can induce the heme oxygenase 1-carbon monoxide (HO-CO) system, which might contribute to systemic hyperperfusion and to regional hyperperfusion of the kidney and other vascular beds via CO-driven vasodilatation [47].
It should be noted that other types of chronic hemolytic anemia such as beta (β)-thalassemia [48,49,50,51] and hemolytic uremic syndrome [52] have been associated with GHF. In β-thalassemia, it has been suggested that GHF could be a consequence of chronic anemia and the iron deposition on the internal milieu within the glomeruli [49, 50].
Autosomal dominant polycystic kidney disease (ADPKD)
This is a common genetic kidney disease affecting 1 in 400 to 1 in 1000 people [53], and it is the primary genetic cause of CKD and kidney failure in the adult population [54]. ADPKD is caused by mutations in either polycystin1 or polycystin2, the proteins that regulate the morphologic configuration of epithelial cells [55]. These mutations lead to the enormously enlarged kidneys caused by the sustained expansion of large numbers of fluid-filled cysts, which are derived from abnormal proliferation of tubular epithelial cells [55, 56].
In patients with ADPKD, the kidney manifestation usually begins at birth with cystic growth and kidney enlargement [8]. These cysts can develop in a minority of medullary and cortical tubules, enlarge exponentially, and compress adjacent parenchyma. This eventually leads to apoptosis, atrophy, and fibrosis of normal functioning parenchyma and the subsequent loss of kidney function [56]. Although the kidney function in individuals with ADPKD usually remains stable until the fourth or fifth decade of life [57], it has been suggested that the preservation of the kidney in the early-stage of the disease is caused by compensatory hyperfiltration of the remnant nephrons [57], which is presented in 21–32% of the pediatric population [53, 58, 59]. Indeed, enlarged kidneys in children with ADPKD have been reported by Wong et al. [59] to be associated with a significantly elevated GFR (which may represent GHF) in comparison to age-matched controls.
Despite the fact that GHF has been considered as one of the markers of ADPKD [60], the exact mechanism by which GHF is induced in children with ADPKD still remains incompletely understood. Nevertheless, Helal et al. reported that the presence of GHF is associated with an increased rate of kidney enlargement over time [53], and this could be driven by the renin–angiotensin–aldosterone system (RAAS), which is known to be stimulated in ADPKD. Indeed, the activation of RAAS raises the angiotensin II (ANG II) level that subsequently increases the glomerular efferent arteriole resistance leading to GHF [53, 61]. In addition, ANG II can enhance cell proliferation, inflammation, oxidant injury, and fibrosis, thus contributing to the growth of the kidney cysts [61]. Furthermore, vasopressin, which is also elevated in patients with ADPKD [58], is reported to play an indirect role on the GHF by reducing sodium concentration at the macula densa, leading to the inhibition TGF control of the GFR [58, 62, 63].
Duchenne muscular dystrophy (DMD)
DMD is an X-linked recessive muscle disorder affecting approximately 1 in 3500 to 1 in 6000 newborn males [64]. This disease is caused by the absence or reduced expression of dystrophin, thereby resulting in a progressive muscle degeneration [65]. There is paucity of data regarding the involvement of the kidney in DMD. A Japanese group reported that kidney failure is a cause of death in 14% of their adult patients with DMD [66] and that 30% of the patients above the age of 30 years had increased plasma levels of cystatin C [67]. Furthermore, in the pediatric population, Braat et al. reported the presence of GHF (measured GFR > 150 mL/min/1.73 m2) using chromium-51-labeled ethylenediamine tetraacetic acid (51Cr-EDTA), in 25% of their patients with DMD [68]. The authors hypothesized that an activated RAAS may act as a possible underlying pathophysiological mechanism due to efferent arteriolar vasoconstriction, a consequence of the low kidney sodium excretion in some patients. To further support the possible role of RAAS activation, more than 50% of the patients in their cohort had elevated blood pressure (BP) and the presence of a non-dipping BP [68].
Although it is still unknown whether mutations in dystrophin protein are responsible for the GHF rather than a secondary cause for the activation of RAAS, the expression of non-muscular isoforms of dystrophin demonstrated in the macula densa, mesangial, and endothelial cells of the kidney might support this possibility [69, 70]. Therefore, future studies should be implemented to shed more insights into the pathophysiology of the kidney damage in patients with DMD.
Diabetes mellitus (DM)
This is a chronic metabolic disease affecting both adult and pediatric populations [71], and it is one of the leading causes of kidney failure and dialysis in western countries. Kidney dysfunctions have been demonstrated to be associated with both type 1 DM (T1DM) and type 2 DM (T2DM), with the pediatric populations with T1DM or T2DM presenting markers of early diabetic kidney disease, such as moderate albuminuria and GHF. GHF is considered as a strong risk factor for progression to CKD and kidney failure and may also predict progressive diabetic kidney disease (DKD) prior to the loss of kidney function [72]. Individuals with diabetes frequently have a significantly higher GFR than nondiabetic counterparts [72, 73], with the prevalence of GHF ranging between 13–52% and 7–40% in pediatric populations with T1DM [73,74,75,76,77] and T2DM [78,79,80,81], respectively.
The potential mechanisms leading to the development of GHF in patients with diabetes remain incompletely unraveled. Several studies in human, particularly in the adult population, and animal models have resulted in several hypotheses. These hypotheses can be grouped into three: ultrastructural changes, vascular theory, and tubular theory [14].
GHF in diabetes has been associated with nephromegaly [82, 83]. Hypertrophy, hyperplasia of the cortical tubuli, and concomitant kidney enlargement are the earliest structural changes in diabetic kidneys. Although there is a glomerular increase in volume, most of the increase in the cortical mass may be due to the hypertrophy of the proximal tubule, which is suggested to be driven by tubular glucose reabsorption, associated with hyperglycemia, thereby leading to increased expression of growth factors such as transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), and insulin-like growth factor (IGF) [84]. In support of this proposed ultrastructural change mechanism, nonspecific and selective sodium glucose transporter-2 (SGLT-2) inhibitors have been shown to attenuate kidney hypertrophy associated with experimental diabetes [85].
Primary abnormalities in vascular control leading to kidney vasodilation and increased kidney blood flow have been observed both in human studies of early diabetes and in animal models [86,87,88,89,90,91]. Animal studies indicate that afferent glomerular arterioles dilate more than the efferent arterioles, thereby raising the GFR, intraglomerular pressure, and filtration fraction [92]. Indeed, in clinical studies, increased GFR and kidney blood flow have been reported in newly diagnosed patients with T1DM and T2DM, thus indicating afferent kidney vasodilation; moreover, patients presented glomerular hypertension as indicated by an elevated filtration fraction [86,87,88,89, 91]. Despite the fact that the mechanism leading to the afferent glomerular vasodilation still remains unclear, elevated levels of insulin, IGF-1, atrial natriuretic peptide (ANP), advanced glycation end products, and increased intrarenal NO signaling have been suggested to contribute to the afferent glomerular vasodilation [8].
Regarding the tubular theory, some investigators have suggested that impairment of the TGF mechanism is the mechanism leading to the development of GHF. Under chronic hyperglycemic conditions in experimental diabetes, the increased proximal tubular reabsorption of glucose and sodium by SGLT-2 causes the inhibition of TGF, via decreased sodium chloride delivery to the macula densa. This reduced delivery of sodium chloride to the macula densa inhibits the conversion of adenosine triphosphate to adenosine, thereby leading to a lower level of adenosine, a vasoconstrictor, and an inadequate afferent arteriolar tone, which eventually results in an increased kidney perfusion [93, 94]. This proposed mechanism has been supported by clinical studies, which showed an enhanced proximal tubule reabsorption of sodium in adult patient populations with T1DM [95] and T2DM [96]. In contrast, a study in mice deficient in the adenosine receptor A1, which lack a TGF mechanism, demonstrated the presence of diabetes-induced GHF [97], thus suggesting that other pathways might be involved in the development of GHF. Therefore, further studies are needed to determine and validate the exact mechanisms of diabetes-induced GHF in humans, and these studies should include the pediatric population.
Malnutrition and obesity
The World Health Organization (WHO) [98] defines malnutrition as “deficiencies or excesses in nutrient intake, imbalances of essential nutrients or impaired nutrient utilization.” Therefore, malnutrition can range from undernutrition, such as stunting, wasting, underweight, and micronutrient deficiencies or insufficiencies, to overweight and obesity as well as diet-related noncommunicable diseases, e.g., DM, cancer, heart disease, and stroke. Children with malnutrition are at increased risk for developing several comorbidities during adulthood, including kidney diseases [99].
Childhood obesity has become a worldwide epidemic [100]. According to the Centers for Disease Control and Prevention (CDC) website, the incidence of obesity in children and adolescents living in the United States of America (USA) is 1 in 5 [101]. The kidney is one of the main target organs for obesity-related health disease, and obesity is an independent risk factor for development of kidney disease. A well-characterized kidney complication associated with obesity is obesity-related glomerulopathy (ORG) [102], and it results from hemodynamic changes, manifesting as glomerular hyperperfusion and GHF [103].
GHF is the earliest manifestation of ORG with a prevalence ranging from 7–31% [104,105,106,107,108,109] in childhood obesity. In these individuals, the increased body mass itself has been suggested as the main driver of GHF [103]. Obesity is associated with an increase in absolute mass of adipose tissue, which leads to the physiological maladaptation of increased cardiac output to compensate for increased blood flow to both adipose and non-adipose tissues, including the kidneys [110]. In the kidneys, this increased cardiac output causes a hemodynamic response at the single-nephron level, resulting in glomerular hypertension and increased capillary hydraulic pressure [111, 112]. Although the exact mechanisms by which obesity alters the kidney hemodynamics at the single-nephron level remain unclear, D’Agati et al. proposed 2 concepts to explain GHF in ORG [113].
In the first concept, the authors proposed that the “primary hemodynamics hypothesis”, in which the primary event is vasodilation of the afferent arterioles, leads to GHF in subjects with obesity. The other proposed concept is “tubulocentric hypothesis” in which the primary event is increased proximal reabsorption of sodium and water by the proximal tubules, thereby leading to decreased solute delivery to the macula densa, TGF deactivation, preglomerular vasodilation, and consequent GHF [113].
It is worth mentioning that several studies have reported that obesity-associated GHF can improve after substantial weight loss [114,115,116], indicating that GHF in obesity represents a reversible physiologic adaptation. Although high-protein diets have been suggested as an effective means for rapid weight loss [117], clinical data have shown an association between GHF and high protein consumption [118]. Therefore, high-protein diets should be avoided, if possible, and plant-based proteins may be recommended for weight loss.
Solitary functioning kidney (SFK)
Individuals with only one functioning kidney have an increased risk for developing CKD in the later life. In children, SFK can either be from a primary (congenital) origin (pSFK) or secondary (acquired) after birth (sSFK) [119]. Two main abnormalities in the spectrum of congenital anomalies of the kidney and urinary tract (CAKUT) underline the pSFK: unilateral multicystic dysplastic kidney (MCDK), and unilateral kidney agenesis (UKA). MCDK, which affects 1 in 4300 newborns [120], is a condition in which the kidney parenchyma shows dysplastic and cystic differentiation with a normally atretic ureter due to abnormal early kidney development [119]. On the other hand, UKA comprises the complete absence of developing kidney tissue and a ureter in fetal life [119], and this usually affects 1 in 2000 newborns [121]. On the contrary, sSFK is acquired from uninephrectomy, performed because of kidney malignancy (e.g., Wilms’ tumor) or after complete loss of kidney function due to recurrent urinary tract infection in the kidney with underlying anomalies such as dysplasia, severe vesicoureteric reflux or obstructive nephropathy, or due to a vasculopathy or trauma [119, 122].
It is well known that reduction in the kidney mass either congenitally or acquired after unilateral nephrectomy in both pediatric and adult populations leads to compensatory kidney growth, which encompasses the hypertrophy of both the kidney tubules and the glomerulus [123]. In pSFK, especially in 24–48% of MCDK cases [120], the compensatory nephron formation which occurs during nephrogenesis may cause the remaining kidney to show hypertrophic growth in utero as early as 20 weeks of gestation [123, 124]. Although there is currently almost no technological technique in clinical settings to detect the number of nephrons in living subjects, an 80% and 56% increase in kidney weight and number of nephrons, respectively, has been reported in a single human case of pSFK compared with age-matched control [125]. Moreover, various animal models of kidney mass reduction during nephrogenesis have reported a compensatory nephron formation, with a 4–50% increase in the number of nephrons [126,127,128]. The underlying stimulus and mechanisms mediating kidney hypertrophy in SFK remain unclear. However, increase in the number of medullary papillae [127, 129], NO system, the renal sympathetic nerves, and the RAAS have been implicated in the compensatory nephrogenesis [123].
The other responses accompanying loss of kidney mass include an increase in single-nephron GFR (SNGFR) and in size and function of kidney tubules (increases in both the length and diameter of the proximal and distal tubules and density of the sodium transporters), thus facilitating a greater reabsorption of the increased filtered load [123, 130,131,132]. This increase in the filtration and function of the kidney tubules helps in compensating for the total GFR in the short term but may also drive glomerular injury and contribute to progressive loss of kidney function in the long run [123, 133]. Thus, GHF is considered as the mechanism of kidney injury in patients with an SFK. A recent meta-analysis of kidney injury after nephrectomy in childhood estimated the proportion of children with SFK who develop kidney injury, as defined by proteinuria, high blood pressure, and a decreased estimated GFR (eGFR), to be 15.3%, 14.5%, and 11.9%, respectively [134]. This clearly indicates that children undergoing a nephrectomy need a long-term follow-up. Therefore, a standardized - follow-up protocol for these children is needed, with an annual estimation of blood pressure and proteinuria, and estimation of GFR once every 5 years, except in conditions of rapid pubertal growth, pregnancy, or obesity, where it should be more frequent [133].
The mechanisms linking GHF to glomerular injury and glomerulosclerosis in SFK are still under investigation, but they implicate a role for podocyte loss in glomerular injury. GHF is influenced by various other intrarenal mechanisms, such as glomerular capillary pressure, single-nephron plasma flow, and TGF [123, 135]. The rise in the SNGFR and the increase in glomerular capillary pressure in remnant nephrons in conditions like SFK have been associated with a reduction in afferent arteriole resistance and resetting of the TGF, thereby exposing podocytes to stretch, tensile stress, and fluid flow shear stress (FFSS), which may over time cause a decrease in the integrity of the glomerular filtration barrier and result in the damage and loss of the podocytes [123].
Factors modulating the development of GHF
The factors contributing to the development of GHF in childhood are very different from those in adults, and among others, they include low birth weight (LBW), gender, ethnicity, elevated blood pressure, inflammation, age, and apolipoprotein L1 (APOL1) risk variants (RVs).
-
a.
Low birth weight
LBW, defined as birth weight below 2500 g, is associated with kidney diseases in adulthood [136,137,138]. LBW is a result of either prematurity or intrauterine growth restriction (IUGR) [137], both of which lead to a congenital lower number of nephrons, an increase in glomerular size as a result of compensatory hypertrophy, and hyperfiltration in the remaining nephrons [136, 137, 139]. In a series of 6 patients (2 women and 4 men, mean age of 32 years), with a history of prematurity and very LBW, presenting persistent proteinuria, kidney histopathology revealed secondary focal segmental glomerulosclerosis (FSGS), without other known risk factors. This small study further reported that prematurity and LBW were associated with glomerulosclerosis and GHF, as a result of adaptive response to an increased hemodynamic stress in a setting of lower number of nephrons, leading to secondary FSGS [137, 140]. More so, Kaze et al. investigated the relationship between birth weight and eGFR in 80 children aged 5–10 years. These authors found a trend towards an increased eGFR (eGFR ≥ 120 mL/min/1.73 m2), although not statistically significant, in children with LBW when compared with children with normal birth weight (NBW) [141].
Compensatory GHF is driven by changes in glomerular hemodynamics and intraglomerular pressure leading to an increase in SNGFR [138, 142, 143]. Similar changes have been demonstrated in animal models of kidney ablation and underline the “GHF theory” [138, 142, 144]. This theory suggests that either a congenital or an acquired reduction in nephron number results in increased glomerular capillary pressure and plasma flow rates causing subsequent increase in SNGFR [92, 143, 144]. Furthermore, this compensatory GHF is an important contributor to progressive kidney damage and kidney failure later in life [136, 138]. In 2 adolescents born extremely premature who developed proteinuria, Hibino et al. found kidney pathological features consistent with glomerular hypertension. The authors suggested that proteinuria seen in these patients was caused by glomerular hypertension and GHF [145]. Similar investigation by Kaze et al. reported significant proteinuria in children with LBW in comparison with those with NBW and related this finding to GHF theory [141].
-
b.
Gender
Gender differences have been reported as a risk factor for GHF in some specific diseases, particularly SCD and DM. In SCD, previous studies in humans as well as in animals have reported an association between male gender and the onset of GHF [29, 146, 147]. In a cohort of 326 children with sickle cell anemia (SCA) living in the Democratic Republic of the Congo (DRC), the authors observed an association between male gender and GHF [9]. This finding is similar to that observed in an adolescent SCD cohort living in Cameroon [29]. Intriguingly, Derebail et al. found that male gender was associated with lower odds of baseline GHF in a cohort of 292 adult patients with SCD [148].
An association between male gender and GHF has been further studied in animal models of SCD. In a humanized sickle cell mice model, Kasztan et al. observed a rapid onset of GHF as well as a strong association between the magnitude of GHF and the degree of long-term kidney injury in male SCA mice [146]. The mechanisms by which gender influences the development of GHF in SCD have not been fully elucidated. However, Kasztan and colleagues postulated, using humanized sickle cell mice, that a greater degree of hypoxia and testosterone level in males, lower hemolysis markers, and a higher fetal hemoglobin level in females might contribute to the gender differences associated with GHF. Indeed, testosterone has been found to increase susceptibility to hemolysis, aggravate ischemia–reperfusion injury, and inhibit NO production, which subsequently could exacerbate the development of kidney injury in males, including GHF [146, 147]. The authors further suggested that these gender differences can be driven by an elevation in endothelin-1 (ET-1) expression leading to changes in the filtration barrier components, which results in GHF [147]. This elevation in ET-1 was significantly greater in male mice when compared with female. In fact, sex hormones have been suggested to modulate the expression of ET-1 [149, 150]. Ovarian hormones, particularly estrogen agonists, have been shown to produce similar effects to those of endothelin-1 type A receptor (ET-A) antagonists, explaining the relative protective advantage in females [146, 147]. Moreover, testosterone may promote kidney injury by upregulating ET-1 production [151].
Contrary to nondiabetic kidney disease, female patients with DM have a greater risk to develop GHF [152]. In a study on adolescents with T2DM aged 12–17 years, girls had a higher baseline eGFR and a greater risk to develop GHF over 5 years compared with boys [79, 153]. Similar findings were observed in a cohort of 98 adolescents with T1DM [74]. Despite these findings, the mechanisms responsible for this gender difference have not been well established [152, 153]. In T1DM, higher efferent arteriolar resistance has been found in women who developed GHF and has been suggested to explain this gender difference in GHF, by increasing intraglomerular pressure [152, 153].
In SFK, gender has been implicated to be associated with the clinical manifestations of GHF. The role of gender is yet to be confirmed in the human population, but animal models of SFK have reported that male animals show a higher degree of GHF [154], a higher blood pressure [132], and a higher glomerular pressure and have more glomerular hypertrophy [155] than their female counterparts. This gender difference has been further explained to be driven by testosterone [155, 156]. Therefore, future studies should confirm this gender association in humans, especially in adolescents and young adults, since the differences between genders only starts to appear from puberty [124].
-
c.
Race/ethnicity
Prior studies in the USA in adolescents and young adults have found an association between race and GHF [10, 105]. Indeed, among nondiabetic adolescents aged 12–17 years in the USA, a variation in the prevalence of GHF according to race has been reported. Hispanic adolescents presented a significantly higher prevalence of GHF compared with other ethnic groups (non-Hispanic Whites and Blacks) [10]. Similarly, Turer et al. observed a racial difference in the prevalence of GHF among US adolescents and young adults with overweight and obesity. Non-Hispanic White adolescents had lower odds to present GHF compared with other ethnic groups (Hispanics, non-Hispanic Blacks, other non-Hispanics [105]). There are a few possible explanations for this racial discrepancy. Socio-economic, behavioral, and genetic risk factors may contribute to the residual confounding unaccounted for. For example, sickle cell trait and RVs in APOL1 gene, which are highly prevalent in African ancestry, have been implicated to contribute to racial discrepancy associated with kidney disease in the African population [10, 157].
-
d.
Apolipoprotein L1 risk variants
APOL1 is a 14.5-kb gene with seven exons encoding APOL1, a 398 amino acid protein. Two main RVs have been identified: G1 (rs73885319, p. serine 342 glycine and rs60910145, p. isoleucine 384 methionine) and G2 (rs71785313, asparagine 388, and tyrosine 389 deletion). These RVs are exclusively found in people of African origin, providing innate protection against human African trypanosomiasis (HAT), the deadliest form of African sleeping sickness. On the other hand, these RVs increase the risk of developing various progressive CKD in people of African ancestry [157]. In children, especially those affected by SCD, human immunodeficiency virus (HIV), or various glomerular diseases, APOL1 RVs have been associated with the development of hypertension, albuminuria, and more rapid decline of kidney function [158, 159]. Studies investigating the association between APOL1 RVs and the occurrence of GHF are limited. However, a recent study in 326 children with SCA from the DRC reported a strong association between APOL1 RVs and GHF in these children. Indeed, the presence of APOL1 RVs was associated with sevenfold increased odds of GHF (adjusted p-value = 0.001) [9].
Although the exact involvement of APOL1 in GHF is still yet to be elucidated, the expression of APOL1 G1 transgene in nephrocytes has been reported to enhance nephrocyte function, causing hypertrophy [160]. Likewise, overexpression of APOL1 in podocyte models has been reported to lead to podocyte detachment, with podocyte expressing APOL1 G2/G2 genotype exhibiting significantly reduced and disorganized actin filaments, and fewer adhesion sites leading to higher permeability to albumin [161]. More so, low nephron endowment is reported to be related to APOL1 RVs, thereby suggesting that GHF might serve as a compensatory adaptation in individuals with APOL1 RVs, as in the case of SFK [6, 162]. Regardless, further studies are still needed to better elucidate the potential role of APOL1 in the development of GHF in children.
-
e.
Elevated blood pressure (hypertension)
In the pediatric population, the presence of elevated blood pressure or hypertension is relatively low, with a prevalence ranging from 2–4% [163, 164]. However, hypertension is not infrequent in children with diseases such as SCD [33, 165], DM [79, 166] and SFK [119, 167], where it has been suggested to play a role in progression of kidney damage.
Nearly three decades ago, GHF was described in patients with essential hypertension and emerged as an early marker of hypertensive nephropathy [168]. In this study, the authors suggested that GHF is driven by increases in intraglomerular pressure as a result of increased efferent arteriolar resistance [169]. In pediatric patients with SCD, GHF has been demonstrated to be associated with elevated blood pressure [21]. Here, the authors reported a positive correlation between GFR and systolic blood pressure, even after adjusting for age, gender, and height. However, it remains unknown whether hypertension is a trigger of GHF or whether hypertension is a consequence of GHF in this disease. Thus, further longitudinal studies should be established to understand this causal relationship in various clinical conditions associated with GHF.
-
f.
Age
Age has been reported as a factor modulating GHF in several clinical conditions. In SCD, age has been reported to have an inverse association with GHF or eGFR [29, 148]. Previous studies have demonstrated an increase in GFR in infancy, which plateaus during adolescence and finally begins to decline thereafter [21, 22]. This decline in GFR after adolescence might represent the actual loss of kidney function [148], as illustrated by an increased prevalence of albuminuria with increasing age in individuals with SCD [29].
Age has also been reported to be an important factor in the clinical manifestation of GFR in SFK. The age at which kidney mass reduction occurs may strongly influence compensatory nephron formation and GFR, with data suggesting that the compensation is more robust when the kidney mass is reduced congenitally or during a young age compared with adults [170]. Indeed, following uninephrectomy in adults, GFR recovers to approximately 70% of pre-uninephrectomy values [171, 172], whereas in children with either congenital or early-sSFK, GFR is preserved at a normal two-kidney level [173]. An explanation for this difference between pSFK and sSFK acquired later in life is that compensatory nephron formation begins earlier in fetal life and continues to progress throughout childhood, while uninephrectomy later in life initiates the remnant nephrons to undergo compensatory changes, by increasing their size (but not their number) in order to mitigate the substantial loss in the filtration surface area [173].
-
g.
Inflammation
The role of inflammation has been generally described in the development of kidney disease. In the pediatric population, inflammatory markers have been implicated to be associated with the development of GHF, particularly in DKD and ORG.
The pathophysiological implications of inflammation in DKD have been described at various levels [174]. A study of adolescents with T1DM showed that GHF was associated with increased excretion of urinary cytokines and chemokines [75]. Indeed, high serum levels of tumor necrosis factor α (TNFα) have been demonstrated to be important in the development of kidney hypertrophy and GHF [175]. To support the role of TNFα in kidney hypertrophy and GHF, urinary TNFα has been reported to be associated with increased sodium retention [176], and this in turn can induce the expression of TGFα and the development of kidney hypertrophy [177].
Emerging evidence from various studies suggest that obesity is a chronic pro-inflammatory disease, with an increasing number of investigations implicating the role of inflammation in the pathogenesis of ORG, including GHF [113, 178]. In a Portuguese cohort of children with overweight and obesity, the authors reported a positive correlation between GFR and myeloperoxidase (MPO) activity [179]. Although the mechanism by which MPO is directly involved in GHF is unknown, the ex vivo perfusion of glomeruli with MPO and hydrogen peroxide has been demonstrated to induce epithelial cell foot process effacement [180] and the local MPO activity has been reported to contribute to glomerular damage [181]. It is worth noting that correlation does not necessarily have to reflect causality, therefore, future studies should shed more insights on such associations.
Also, the adipose tissue can act as an endocrine organ for the secretion of angiogenic and inflammatory adipokines, which might have distant effects in the kidney [113, 182], such as the alteration of the glomerular cell size and hypertrophy in glomerular mesangial cells, thereby leading to GHF [183].
An overview of the factors modulating the development of GHF in the pediatric population is presented in Table 2.
Glomerular hyperfiltration: when is it damaging?
GHF has been associated with progressive kidney injury and loss of kidney function leading to kidney failure [184]. However, the exact mechanisms by which GHF induces this kidney injury are not fully elucidated.
Previous studies have implicated RAAS in the development of GHF-mediated injury via inducing alterations in kidney hemodynamics [184, 185]. This finding justified the use of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) to treat patients with GHF. These drugs have effectively delayed the progression of GHF-mediated injury in some but not all kidney diseases, such as SCD, ADPKD, and CAKUT [184, 185].
Another recently proposed mechanism for GHF-mediated injury is the effect of hyperfiltration-related mechanical forces, namely tensile stress and FFSS on the filtration barrier [186], as illustrated in Fig. 3. Tensile stress results from an increase in glomerular capillary pressure on capillary wall structures (Fig. 3a), leading to podocyte damage through the activation of the RAAS. On the other hand, FFSS results from a high ultrafiltrate flow in the Bowman’s space (Fig. 3a), causing podocyte and tubular damage by activating the cyclooxygenase 2–prostaglandin E2–E-prostanoid 2 receptor (COX2–PGE2–EP2) pathway. Indeed, FFSS upregulates COX2, which subsequently activates arachidonic acid metabolism, resulting in increased synthesis of prostaglandins, especially PGE2, a ligand of EP2 receptor [185]. Elevated PGE2 levels act as a mediator of GHF-mediated injury in podocytes and tubules [184].

Proposed mechanism of glomerular hyperfiltration-related kidney damage. Glomerular hyperfiltration is associated with two mechanical forces in the filtration barrier, namely tensile stress (TS) and fluid flow shear stress (FFSS). a Increased glomerular capillary pressure (PCG) causes TS on the capillary wall structure and podocyte, while increased single-nephron glomerular filtration rate (SNGFR) leads to high ultrafiltrate flow in Bowman’s space, exerting a fluid flow shear stress (FFSS) on the podocyte. b These two mechanical forces can cause damage to kidney cells through different pathways. TS activates the renin–angiotensin–aldosterone system (RAAS), whereas FFSS activates the cyclooxygenase 2–prostaglandin E2–E-prostanoid 2 receptor (COX2–PGE2–EP2) axis. Subsequently, these forces cause podocyte and tubular damage, resulting in proteinuria, glomerulosclerosis, tubulointerstitial inflammation, and fibrosis. All these detrimental factors can induce progressive kidney injury and loss of kidney function leading to kidney failure. SNGFR, single-nephron glomerular filtration rate; GFR, glomerular filtration rate; FFSS, fluid flow shear stress; TS, tensile stress; PCG, glomerular capillary pressure; RAAS, renin–angiotensin–aldosterone system; COX2–PGE2–EP2, cyclooxygenase 2–prostaglandin E2–E-prostanoid 2 receptor
In podocyte, these biochemical forces cause damage to podocyte structure and function, leading to an increased glomerular basement membrane length and surface. As a result, a mismatch develops between the glomerular surface area and the podocyte foot process coverage. Podocyte reacts via cellular hypertrophy to cover this increased glomerular surface. Thus, the podocyte’s body becomes stretched and thinned with foot processes deformed [184, 186]. All these changes might result in podocyte detachment, increased glomerular protein permeability, and glomerulosclerosis [184].
Interestingly, the effects of podocyte hypertrophic stress in the development and progression of CKD have been studied in diabetic and nondiabetic rat models subjected to high nutrient intake [187, 188]. In these studies, an increased release of growth factors and exposure to abundant nutrients was reported to trigger podocyte hypertrophic stress via glomerular volume enlargement and activation of the mammalian target of rapamycin complex 1 (mTORC1) pathway. In turn, these led to accelerated podocyte detachment, reduced podocyte density, albuminuria, glomerulosclerosis, and ultimately progression to kidney failure. Moreover, the authors demonstrated potential renoprotective effects of nutritional reduction in ameliorating kidney injury induced by podocyte hypertrophic stress [187, 188]. As previously mentioned, this approach has been shown to be effective in the management of GHF in obesity.
Likewise, these forces induce tubular damage characterized by dilatation of tubular urinary spaces, epithelial cell hypertrophy, and increased proximal tubular sodium reabsorption, thereby resulting in tubulointerstitial inflammation, hypoxia, and fibrosis [184, 186] (Fig. 3b). Therefore, the COX2–PGE2–EP2 pathway may be a potential target for developing novel therapies against GHF-mediated injury [185].
Despite these findings, studies investigating the long-term outcomes of GHF in children are lacking. In a humanized sickle cell mice model, Kasztan et al. found that early GHF was a determinant and a predictor of subsequent progression to CKD, only in the male mice [146]. Indeed, in these male mice, GHF developed at 12 weeks of age and was significantly correlated with kidney damage (manifesting as proteinuria, albuminuria, decline of GFR, and elevated plasma creatinine) at 32 weeks. In addition, these investigators found that the magnitude of GHF was associated with the degree of subsequent kidney damage [146]. In a cohort of a pediatric population with SCA, Lebensburger et al. conducted a prospective cohort study to determine the relationship between GHF and the development of albuminuria [28]. These investigators found that children who presented GHF during early childhood were more likely to develop persistent albuminuria at an earlier age than those without GHF. These data suggested that GHF precedes the development of persistent albuminuria in children with SCA. Nevertheless, more prospective studies are needed to better evaluate the long-term consequences of GHF in patients with SCD.
In patients with DM, data regarding the association of GHF and adverse kidney outcomes are controversial. Prior studies suggested that GHF was associated with an increased risk of developing adverse kidney outcomes in patients with T1DM and T2DM [189, 190]. On the contrary, in a cohort study of 446 participants with T1DM who underwent measurement of GFR using 125I-iothalamate clearance, 106 (24%) presented GHF at baseline. Over a median follow-up of 28 years, 53 (12%) participants presented decreased eGFR (< 60 ml/min per 1.73 m2). However, the investigators did not find an association between early GHF and the risk of developing decreased eGFR or severely increased albuminuria (albuminuria excretion rate ≥ 300 mg/24 h) in the multivariable-adjusted Cox proportional hazards model, suggesting that GHF is not a marker of adverse kidney outcomes in patients with T1DM [191]. It is notable that this study provided robust data on the long-term outcomes of GHF in T1DM patients, with a longer follow-up duration and a gold standard measurement of GFR, offering an optimal classification of GHF at baseline [1].
Future perspectives and closing remarks
A supraphysiological elevation of GFR might occur in the pediatric population, especially in those with clinical conditions, such as SCD, DM, ADPKD, obesity, and DMD. In children with reduced numbers of functioning nephrons, elevated GFR remains the main compensatory mechanism. Nevertheless, GHF displays different etiologies and may cause CKD in people. It is therefore important to unravel the molecular mechanisms responsible for these features in order to find new targets for future therapy which can be used to identify and monitor the disease progression and outcome in high-risk individuals. For this purpose, research in identification of novel prognostic indicators (biomarkers) of disease progression is needed to detect those individuals who would benefit from early-life treatment before the onset of kidney injury. Thus, large cohort studies in the pediatric population are required to help identify these possible prognostic biomarkers. Several interventions like NO bioavailability, SGLT-2 inhibitors, and RAAS blockade are associated with ameliorating GHF in some clinical conditions. The potential benefit of these candidate treatments in other diseases associated with GHF should be tested in available disease-specific model systems, as well as in clinical trials.
Key summary points
-
GHF is generally defined as a supraphysiologic increase of GFR that can occur from an increase in factors determining GFR, such as kidney plasma flow, hydraulic pressure across the kidney membrane, or an ultrafiltration coefficient.
-
The exact threshold for defining GHF is not well established in pediatric population, and it ranges from 120–180 ml/min per 1.73 m2.
-
GHF can occur in either physiologic or pathologic states. In physiologic state, it can occur after consumption of high protein meals or during pregnancy. In pathologic states, GHF can be due to various diseases which are either congenital or acquired. These pathological conditions include SCD, ADPK, DMD, DM, obesity, and SFK.
-
The pathophysiological mechanisms mediating GHF vary between different clinical conditions, occurring either at the single-nephron level or in the whole-kidney.
-
The development of GHF in children can be mediated via multiple factors that differ from those seen in the adult population.
-
GHF can induce progressive kidney injury and loss of kidney function leading to kidney failure through the activation of RAAS and the direct effect of hyperfiltration-related mechanical forces.
Multiple-choice questions
-
1.
Which factor determines GFR?
-
a)
Permeability of glomerular basement membrane
-
b)
Hydraulic pressure
-
c)
Fluid flow shear stress
-
d)
Tensile stress
-
e)
COX2–PGE2–EP2 pathway
-
a)
-
2.
Which of the following statements is FALSE?
-
a)
The exact threshold for defining GHF can range from 120–180 ml/min per 1.73 m2
-
b)
GHF can occur at both the single-nephron and the whole-kidney levels
-
c)
The mechanisms mediating GHF are similar in all clinical conditions
-
d)
In SFK, GHF is considered as the main mechanism of kidney injury
-
e)
The etiology of GHF can be multifactorial
-
a)
-
3.
Which one of these clinical conditions is not linked with GHF in the pediatric population?
-
a)
Sickle cell disease
-
b)
Renal agenesis/aplasia
-
c)
Enuresis
-
d)
Obesity
-
e)
Diabetes mellitus
-
a)
-
4.
Which clinical condition is associated with single-nephron GHF?
-
a)
Duchenne muscular dystrophy
-
b)
Autosomal dominant polycystic kidney disease
-
c)
Sickle cell disease
-
d)
Enuresis
-
e)
Cancer
-
a)
-
5.
Which of the following hormones plays a role in the association between gender and GHF?
-
a)
Gastrin
-
b)
Insulin
-
c)
Cortisol
-
d)
Progesterone
-
e)
Testosterone
-
a)
References
Tummalapalli SL, Shlipak MG (2019) Hyperfiltration: much ado about nothing? Clin J Am Soc Nephrol 14:789–791
Oh SW, Yang JH, Kim MG, Cho WY, Jo SK (2020) Renal hyperfiltration as a risk factor for chronic kidney disease: a health checkup cohort study. PLoS One 15:e0238177
Cachat F, Combescure C, Cauderay M, Girardin E, Chehade H (2015) A systematic review of glomerular hyperfiltration assessment and definition in the medical literature. Clin J Am Soc Nephrol 10:382–389
Park M, Yoon E, Lim YH, Kim H, Choi J, Yoon HJ (2015) Renal hyperfiltration as a novel marker of all-cause mortality. J Am Soc Nephrol 26:1426–1433
Kanbay M, Ertuglu LA, Afsar B, Ozdogan E, Kucuksumer ZS, Ortiz A, Covic A, Kuwabara M, Cherney DZI, van Raalte DH, de Zeeuw D (2019) Renal hyperfiltration defined by high estimated glomerular filtration rate: a risk factor for cardiovascular disease and mortality. Diabetes Obes Metab 21:2368–2383
Cortinovis M, Perico N, Ruggenenti P, Remuzzi A, Remuzzi G (2022) Glomerular hyperfiltration. Nat Rev Nephrol 18:435–451
Premaratne E, Macisaac RJ, Tsalamandris C, Panagiotopoulos S, Smith T, Jerums G (2005) Renal hyperfiltration in type 2 diabetes: effect of age-related decline in glomerular filtration rate. Diabetologia 48:2486–2493
Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW (2012) Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol 8:293–300
Adebayo OC, Betukumesu DK, Nkoy AB, Adesoji OM, Ekulu PM, Van den Heuvel LP, Levtchenko EN, Labarque V (2022) Clinical and genetic factors are associated with kidney complications in African children with sickle cell anaemia. Br J Haematol 196:204–214
Lee AM, Charlton JR, Carmody JB, Gurka MJ, DeBoer MD (2017) Metabolic risk factors in nondiabetic adolescents with glomerular hyperfiltration. Nephrol Dial Transplant 32:1517–1524
Pottel H, Bjork J, Delanaye P, Nyman U (2022) Evaluation of the creatinine-based chronic kidney disease in children (under 25 years) equation in healthy children and adolescents. Pediatr Nephrol 37:2213–2216
van Dam M, Pottel H, Vreugdenhil ACE (2022) Creatinine-based GFR-estimating equations in children with overweight and obesity. Pediatr Nephrol 37:2393–2403
Xargay-Torrent S, Puerto-Carranza E, Marcelo I, Mas-Pares B, Gomez-Vilarrubla A, Martinez-Calcerrada JM, de Zegher F, Ibanez L, Lopez-Bermejo A, Bassols J (2021) Estimated glomerular filtration rate and cardiometabolic risk factors in a longitudinal cohort of children. Sci Rep 11:11702
Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, Joles JA (2017) Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol 28:1023–1039
Lumbers ER, Kandasamy Y, Delforce SJ, Boyce AC, Gibson KJ, Pringle KG (2020) Programming of renal development and chronic disease in adult life. Front Physiol 11:757
Ranque B, Menet A, Diop IB, Thiam MM, Diallo D, Diop S, Diagne I, Sanogo I, Kingue S, Chelo D, Wamba G, Diarra M, Anzouan JB, N’Guetta R, Diakite CO, Traore Y, Legueun G, Deme-Ly I, Belinga S, Boidy K, Kamara I, Tharaux PL, Jouven X (2014) Early renal damage in patients with sickle cell disease in sub-Saharan Africa: a multinational, prospective, cross-sectional study. Lancet Haematol 1:e64-73
Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI (2013) Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381:142–151
Adebayo OC, Van den Heuvel LP, Olowu WA, Levtchenko EN, Labarque V (2022) Sickle cell nephropathy: insights into the pediatric population. Pediatr Nephrol 37:1231–1243
Hirschberg R (2010) Glomerular hyperfiltration in sickle cell disease. Clin J Am Soc Nephrol 5:748–749
Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV, Barredo JC, Brown RC, Sarnaik SA, Howard TH, Wynn LW, Kutlar A, Armstrong FD, Files BA, Goldsmith JC, Waclawiw MA, Huang X, Thompson BW (2011) Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 377:1663–1672
Aygun B, Mortier NA, Smeltzer MP, Hankins JS, Ware RE (2011) Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatr Nephrol 26:1285–1290
Yee MM, Jabbar SF, Osunkwo I, Clement L, Lane PA, Eckman JR, Guasch A (2011) Chronic kidney disease and albuminuria in children with sickle cell disease. Clin J Am Soc Nephrol 6:2628–2633
Ranabothu S, Hafeman M, Manwani D, Reidy K, Morrone K, Lorenzo J, Tria B, Kaskel F, Mahgerefteh J (2020) Ambulatory hypertension in pediatric patients with sickle cell disease and its association with end-organ damage. Cureus 12:e11707
Nnaji UM, Ogoke CC, Okafor HU, Achigbu KI (2020) Sickle cell nephropathy and associated factors among asymptomatic children with sickle cell anaemia. Int J Pediatr 2020:1286432
Inusa BPD, Booth C, Iduoriyekemwen N (2020) Glomerular hyperfiltration and albuminuria in adolescents with sickle cell disease: retrospective cross-sectional study. Blood 136(Suppl 1):1–2
Belisario AR, de Almeida JA, Mendes FG, da Silva DMM, Planes W, Rezende PV, Silva CM, Brito AC, Sales RR, Viana MB, Simoes ESAC (2020) Prevalence and risk factors for albuminuria and glomerular hyperfiltration in a large cohort of children with sickle cell anemia. Am J Hematol 95:E125–E128
Zahr RS, Yee ME, Weaver J, Twombley K, Matar RB, Aviles D, Sreedharan R, Rheault MN, Malatesta-Muncher R, Stone H, Srivastava T, Kapur G, Baddi P, Volovelsky O, Pelletier J, Gbadegesin R, Seeherunvong W, Patel HP, Greenbaum LA (2019) Kidney biopsy findings in children with sickle cell disease: a Midwest Pediatric Nephrology Consortium study. Pediatr Nephrol 34:1435–1445
Lebensburger JD, Aban I, Pernell B, Kasztan M, Feig DI, Hilliard LM, Askenazi DJ (2019) Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol 94:417–423
Geard A, Pule GD, Chetcha Chemegni B, Ngo Bitoungui VJ, Kengne AP, Chimusa ER, Wonkam A (2017) Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol 178:629–639
Brewin J, Tewari S, Hannemann A, Al Balushi H, Sharpe C, Gibson JS, Rees DC (2017) Early markers of sickle nephropathy in children with sickle cell anemia are associated with red cell cation transport activity. HemaSphere 1:e2
Aloni MN, Ngiyulu RM, Ekulu PM, Mbutiwi FIN, Makulo JR, Gini-Ehungu JL, Nseka NM, Lepira FB (2017) Glomerular hyperfiltration is strongly correlated with age in Congolese children with sickle cell anaemia. Acta Paediatr 106:819–824
Aloni MN, Ngiyulu RM, Gini-Ehungu JL, Nsibu CN, Ekila MB, Lepira FB, Nseka NM (2014) Renal function in children suffering from sickle cell disease: challenge of early detection in highly resource-scarce settings. PLoS One 9:e96561
Bodas P, Huang A, O’Riordan MA, Sedor JR, Dell KM (2013) The prevalence of hypertension and abnormal kidney function in children with sickle cell disease -a cross sectional review. BMC Nephrol 14:2–7
Aygun B, Mortier NA, Smeltzer MP, Shulkin BL, Hankins JS, Ware RE (2013) Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol 88:116–119
Allon M, Lawson L, Eckman JR, Delaney V, Bourke E (1988) Effects of nonsteroidal antiinflammatory drugs on renal function in sickle cell anemia. Kidney Int 34:500–506
Schmitt F, Martinez F, Brillet G, Giatras I, Choukroun G, Girot R, Bachir D, Galacteros F, Lacour B, Grunfeld JP (1998) Early glomerular dysfunction in patients with sickle cell anemia. Am J Kidney Dis 32:208–214
Etteldorf JN, Tuttle AW, Clayton GW (1952) Renal function studies in pediatrics. 1. Renal hemodynamics in children with sickle cell anemia. AMA Am J Dis Child 83:185–191
De Jong PE, Van Eps LWS (1985) Sickle cell nephropathy: new insights into its pathophysiology. Kidney Int 27:711–717
Ataga KI, Orringer EP (2000) Renal abnormalities in sickle cell disease. Am J Hematol 63:205–211
Turner N, Lameire N (2016) Oxford textbook of clinical nephrology. Oxford University Press, Oxford
Bernstein J, Whitten CF (1960) A histologic appraisal of the kidney in sickle cell anemia. Arch Pathol 70:407–418
Bhathena DB, Sondheimer JH (1991) The glomerulopathy of homozygous sickle hemoglobin (SS) disease: morphology and pathogenesis. J Am Soc Nephrol 1:1241–1252
Olaniran KO, Eneanya D, Nigwekar SU (2019) Sickle cell nephropathy in the pediatric population. Blood Purif 47:205–213
Sharpe CC, Thein SL (2011) Sickle cell nephropathy - a practical approach. Br J Haematol 155:287–297
Haymann JP, Hammoudi N, Livrozet M, Santin A, Mattioni S, Letavernier E, Frochot V, Jacques CS, Steichen O, Grateau G, Chaignon M, Lionnet F (2021) Hemodynamic and biological correlates of glomerular hyperfiltration in sickle cell patients before and under renin-angiotensin system blocker. Sci Rep 11:11682
Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F (2010) Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol 5:756–761
Nath KA, Katusic ZS (2012) Vasculature and kidney complications in sickle cell disease. J Am Soc Nephrol 23:781–784
Ziyadeh FN, Musallam KM, Mallat NS, Mallat S, Jaber F, Mohamed AA, Koussa S, Taher AT (2012) Glomerular hyperfiltration and proteinuria in transfusion-independent patients with beta-thalassemia intermedia. Nephron Clin Pract 121:c136-143
Mohd Zikre N, Muhamad NA, Eng CSY, Zailanalhuddin NE, Lai CD, Foo JC, Yap SL, Ariffin H, Abu Bakar K (2021) Occult kidney dysfunction in children with transfusion-dependent thalassemia. Front Pediatr 9:754813
Quinn CT, Johnson VL, Kim HY, Trachtenberg F, Vogiatzi MG, Kwiatkowski JL, Neufeld EJ, Fung E, Oliveri N, Kirby M, Giardina PJ, Thalassemia Clinical Research Network (2011) Renal dysfunction in patients with thalassaemia. Br J Haematol 153:111–117
Deveci B, Kurtoglu A, Kurtoglu E, Salim O, Toptas T (2016) Documentation of renal glomerular and tubular impairment and glomerular hyperfiltration in multitransfused patients with beta thalassemia. Ann Hematol 95:375–381
Moghal NE, Ferreira MA, Howie AJ, Milford DV, Raafat E, Taylor CM (1998) The late histologic findings in diarrhea-associated hemolytic uremic syndrome. J Pediatr 133:220–223
Helal I, Reed B, McFann K, Yan XD, Fick-Brosnahan GM, Cadnapaphornchai M, Schrier RW (2011) Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6:2439–2443
Helal I, Reed B, Schrier RW (2012) Emergent early markers of renal progression in autosomal-dominant polycystic kidney disease patients: implications for prevention and treatment. Am J Nephrol 36:162–167
Grantham JJ, Chapman AB, Torres VE (2006) Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol 1:148–157
Grantham JJ, Mulamalla S, Swenson-Fields KI (2011) Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7:556–566
Messchendorp AL, van Londen M, Taylor JM, de Borst MH, Navis G, Casteleijn NF, Gaillard C, Bakker SJL, Gansevoort RT, DIPAK Consortium (2018) Kidney function reserve capacity in early and later stage autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 13:1680–1692
Selistre L, de Souza V, Ranchin B, Hadj-Aissa A, Cochat P, Dubourg L (2012) Early renal abnormalities in children with postnatally diagnosed autosomal dominant polycystic kidney disease. Pediatr Nephrol 27:1589–1593
Wong H, Vivian L, Weiler G, Filler G (2004) Patients with autosomal dominant polycystic kidney disease hyperfiltrate early in their disease. Am J Kidney Dis 43:624–628
Dimitrakov D, Kumchev E, Lyutakova E, Grigorov L (1993) Glomerular hyperfiltration and serum beta 2-microglobulin used as early markers in diagnosis of autosomal dominant polycystic kidney disease. Folia Med (Plovdiv) 35:59–62
Schrier RW (2009) Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 20:1888–1893
Torres VE (2005) Vasopressin antagonists in polycystic kidney disease. Kidney Int 68:2405–2418
Bankir L, Bichet DG (2019) What can copeptin tell us in patients with autosomal dominant polycystic disease? Kidney Int 96:19–22
Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 71:304–313
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Matsumura T, Saito T, Fujimura H, Shinno S, Sakoda S (2011) A longitudinal cause-of-death analysis of patients with Duchenne muscular dystrophy. Rinsho Shinkeigaku 51:743–750
Matsumura T, Saito T, Fujimura H, Sakoda S (2012) Renal dysfunction is a frequent complication in patients with advanced stage of Duchenne muscular dystrophy. Rinsho Shinkeigaku 52:211–217
Braat E, Hoste L, De Waele L, Gheysens O, Vermeersch P, Goffin K, Pottel H, Goemans N, Levtchenko E (2015) Renal function in children and adolescents with Duchenne muscular dystrophy. Neuromuscul Disord 25:381–387
Haenggi T, Fritschy JM (2006) Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell Mol Life Sci 63:1614–1631
Haenggi T, Schaub MC, Fritschy JM (2005) Molecular heterogeneity of the dystrophin-associated protein complex in the mouse kidney nephron: differential alterations in the absence of utrophin and dystrophin. Cell Tissue Res 319:299–313
Amatruda M, Gembillo G, Giuffrida AE, Santoro D, Conti G (2021) The aggressive diabetic kidney disease in youth-onset type 2 diabetes: pathogenetic mechanisms and potential therapies. Medicina (Kaunas) 57:868
Bjornstad P, Cherney DZ, Maahs DM, Nadeau KJ (2016) Diabetic kidney disease in adolescents with type 2 diabetes: new insights and potential therapies. Curr Diab Rep 16:11
Bjornstad P, Roncal C, Milagres T, Pyle L, Lanaspa MA, Bishop FK, Snell-Bergeon JK, Johnson RJ, Wadwa RP, Maahs DM (2016) Hyperfiltration and uricosuria in adolescents with type 1 diabetes. Pediatr Nephrol 31:787–793
Lovshin JA, Skrtic M, Bjornstad P, Moineddin R, Daneman D, Dunger D, Reich HN, Mahmud F, Scholey J, Cherney DZI, Sochett E (2018) Hyperfiltration, urinary albumin excretion, and ambulatory blood pressure in adolescents with type 1 diabetes mellitus. Am J Physiol Renal Physiol 314:F667–F674
Har RL, Reich HN, Scholey JW, Daneman D, Dunger DB, Moineddin R, Dalton RN, Motran L, Elia Y, Deda L, Ostrovsky M, Sochett EB, Mahmud FH, Cherney DZ (2014) The urinary cytokine/chemokine signature of renal hyperfiltration in adolescents with type 1 diabetes. PLoS One 9:e111131
Hernandez-Marco R, Codoner-Franch P, Pons Morales S, Del Castillo VC, Boix Garcia L, Valls Belles V (2009) Oxidant/antioxidant status and hyperfiltration in young patients with type 1 diabetes mellitus. Pediatr Nephrol 24:121–127
Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA (2006) Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol 17:1703–1709
Dart AB, McGavock J, Sharma A, Chateau D, Schwartz GJ, Blydt-Hansen T (2019) Estimating glomerular filtration rate in youth with obesity and type 2 diabetes: the iCARE study equation. Pediatr Nephrol 34:1565–1574
Bjornstad P, Nehus E, El Ghormli L, Bacha F, Libman IM, McKay S, Willi SM, Laffel L, Arslanian S, Nadeau KJ, TODAY Study Group (2018) Insulin sensitivity and diabetic kidney disease in children and adolescents with type 2 diabetes: an observational analysis of data from the TODAY clinical trial. Am J Kidney Dis 71:65–74
Bjornstad P, Maahs DM, Cherney DZ, Cree-Green M, West A, Pyle L, Nadeau KJ (2014) Insulin sensitivity is an important determinant of renal health in adolescents with type 2 diabetes. Diabetes Care 37:3033–3039
Sellers EA, Blydt-Hansen TD, Dean HJ, Gibson IW, Birk PE, Ogborn M (2009) Macroalbuminuria and renal pathology in First Nation youth with type 2 diabetes. Diabetes Care 32:786–790
Osterby R, Gundersen HJ (1975) Glomerular size and structure in diabetes mellitus. I. Early Abnormalities. Diabetologia 11:225–229
Jerums G, Premaratne E, Panagiotopoulos S, MacIsaac RJ (2010) The clinical significance of hyperfiltration in diabetes. Diabetologia 53:2093–2104
Trevisan R, Dodesini AR (2017) The hyperfiltering kidney in diabetes. Nephron 136:277–280
Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T (2014) SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306:F194-204
Bank N (1991) Mechanisms of diabetic hyperfiltration. Kidney Int 40:792–807
Vora JP, Dolben J, Dean JD, Thomas D, Williams JD, Owens DR, Peters JR (1992) Renal hemodynamics in newly presenting non-insulin dependent diabetes mellitus. Kidney Int 41:829–835
Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, Hirschman GH, Myers BD (1996) Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med 335:1636–1642
Keller CK, Bergis KH, Fliser D, Ritz E (1996) Renal findings in patients with short-term type 2 diabetes. J Am Soc Nephrol 7:2627–2635
O’Bryan GT, Hostetter TH (1997) The renal hemodynamic basis of diabetic nephropathy. Semin Nephrol 17:93–100
Levine DZ (2008) Can rodent models of diabetic kidney disease clarify the significance of early hyperfiltration?: recognizing clinical and experimental uncertainties. Clin Sci (Lond) 114:109–118
Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM (1981) Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol 241:F85-93
Vallon V, Richter K, Blantz RC, Thomson S, Osswald H (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V (2001) Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest 107:217–224
Vervoort G, Veldman B, Berden JH, Smits P, Wetzels JF (2005) Glomerular hyperfiltration in type 1 diabetes mellitus results from primary changes in proximal tubular sodium handling without changes in volume expansion. Eur J Clin Invest 35:330–336
Pruijm M, Wuerzner G, Maillard M, Bovet P, Renaud C, Bochud M, Burnier M (2010) Glomerular hyperfiltration and increased proximal sodium reabsorption in subjects with type 2 diabetes or impaired fasting glucose in a population of the African region. Nephrol Dial Transplant 25:2225–2231
Sallstrom J, Carlsson PO, Fredholm BB, Larsson E, Persson AE, Palm F (2007) Diabetes-induced hyperfiltration in adenosine A(1)-receptor deficient mice lacking the tubuloglomerular feedback mechanism. Acta Physiol (Oxf) 190:253–259
World health organization. Health topics. Malnutrition. https://www.who.int/health-topics/malnutrition. Accessed 16 February 2022
Vivante A, Golan E, Tzur D, Leiba A, Tirosh A, Skorecki K, Calderon-Margalit R (2012) Body mass index in 1.2 million adolescents and risk for end-stage renal disease. Arch Intern Med 172:1644–1650
Wang Y, Lobstein T (2006) Worldwide trends in childhood overweight and obesity. Int J Pediatr Obes 1:11–25
Centers for Disease Control and Prevention (2021) Childhood Overweight & Obesity. Overweight & Obesity. https://www.cdc.gov/obesity/childhood/index.html. Accessed 18 February 2022
Okabayashi Y, Tsuboi N, Sasaki T, Haruhara K, Kanzaki G, Koike K, Shimizu A, D’Agati VD, Yokoo T (2020) Single-nephron GFR in patients with obesity-related glomerulopathy. Kidney Int Rep 5:1218–1227
Ruster C, Wolf G (2013) The role of the renin-angiotensin-aldosterone system in obesity-related renal diseases. Semin Nephrol 33:44–53
Muzzio ML, Kabakian ML, Morosan-Allo Y, Ferrari S, Fallahi P, Fernandez J, Santucci MP, Andres-Lacueva C, Antonelli A, Brenta G, Merono T (2020) Association of glomerular hyperfiltration with serum chemokine levels and metabolic features in prepubertal children with overweight/obesity. Nutr Metab Cardiovasc Dis 30:1188–1195
Turer CB, Baum M, Dubourg L, Selistre LS, Skinner AC (2019) Prevalence of hyperfiltration among US youth/young adults with overweight and obesity: a population-based association study. Obes Sci Pract 5:570–580
Santucci MP, Muzzio ML, Peredo MS, Brovarone L, Scricciolo R, Diez C, Andres-Lacueva C, Kabakian ML, Merono T (2020) Different alterations of glomerular filtration rate and their association with uric acid in children and adolescents with type 1 diabetes or with overweight/obesity. Pediatr Diabetes 21:657–663
Arora S, Dunkley L, Waldman LM, Chin VL, Umpaichitra V (2020) Kidney function in minority children and adolescents with metabolically healthy and unhealthy obesity. Clin Obes 10:e12345
Bielopolski D, Singh N, Bentur OS, Renert-Yuval Y, MacArthur R, Vasquez KS, Moftah DS, Vaughan RD, Charytan DM, Kost RG, Tobin JN (2021) Obesity related glomerulopathy in adolescent women: the effect of body surface area. Kidney360 3:113–121
Franchini S, Savino A, Marcovecchio ML, Tumini S, Chiarelli F, Mohn A (2015) The effect of obesity and type 1 diabetes on renal function in children and adolescents. Pediatr Diabetes 16:427–433
Srivastava T (2006) Nondiabetic consequences of obesity on kidney. Pediatr Nephrol 21:463–470
Nehus E, Mitsnefes M (2019) Childhood obesity and the metabolic syndrome. Pediatr Clin North Am 66:31–43
Palatini P (2012) Glomerular hyperfiltration: a marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol Dial Transplant 27:1708–1714
D’Agati VD, Chagnac A, de Vries AP, Levi M, Porrini E, Herman-Edelstein M, Praga M (2016) Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 12:453–471
Nehus EJ, Khoury JC, Inge TH, Xiao N, Jenkins TM, Moxey-Mims MM, Mitsnefes MM (2017) Kidney outcomes three years after bariatric surgery in severely obese adolescents. Kidney Int 91:451–458
van Dam M, Rijks J, Dorenbos E, Horuz F, van Dael K, Vreugdenhil A (2019) The effect of one year lifestyle intervention on eGFR in children and adolescents with overweight, obesity and morbid obesity. Sci Rep 9:4504
Chang AR, George J, Levey AS, Coresh J, Grams ME, Inker LA (2020) Performance of glomerular filtration rate estimating equations before and after bariatric surgery. Kidney Med 2(699–706):e691
Ko GJ, Rhee CM, Kalantar-Zadeh K, Joshi S (2020) The effects of high-protein diets on kidney health and longevity. J Am Soc Nephrol 31:1667–1679
Kamper AL, Strandgaard S (2017) Long-term effects of high-protein diets on renal function. Annu Rev Nutr 37:347–369
Westland R, Schreuder MF, Bokenkamp A, Spreeuwenberg MD, van Wijk JA (2011) Renal injury in children with a solitary functioning kidney–the KIMONO study. Nephrol Dial Transplant 26:1533–1541
Schreuder MF, Westland R, van Wijk JA (2009) Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 24:1810–1818
Westland R, Schreuder MF, Ket JC, van Wijk JA (2013) Unilateral renal agenesis: a systematic review on associated anomalies and renal injury. Nephrol Dial Transplant 28:1844–1855
Narkun-Burgess DM, Nolan CR, Norman JE, Page WF, Miller PL, Meyer TW (1993) Forty-five year follow-up after uninephrectomy. Kidney Int 43:1110–1115
McArdle Z, Schreuder MF, Moritz KM, Denton KM, Singh RR (2020) Physiology and pathophysiology of compensatory adaptations of a solitary functioning kidney. Front Physiol 11:725
Schreuder MF (2018) Life with one kidney. Pediatr Nephrol 33:595–604
Maluf NS (1997) On the enlargement of the normal congenitally solitary kidney. Br J Urol 79:836–841
Westland R, Schreuder MF, van Goudoever JB, Sanna-Cherchi S, van Wijk JA (2014) Clinical implications of the solitary functioning kidney. Clin J Am Soc Nephrol 9:978–986
van Vuuren SH, Sol CM, Broekhuizen R, Lilien MR, Oosterveld MJ, Nguyen TQ, Goldschmeding R, de Jong TP (2012) Compensatory growth of congenital solitary kidneys in pigs reflects increased nephron numbers rather than hypertrophy. PLoS One 7:e49735
Douglas-Denton R, Moritz KM, Bertram JF, Wintour EM (2002) Compensatory renal growth after unilateral nephrectomy in the ovine fetus. J Am Soc Nephrol 13:406–410
Snoek R, de Heus R, de Mooij KJ, Pistorius LR, Lilien MR, Lely AT, Bekker MN, de Jong T (2018) Assessing nephron hyperplasia in fetal congenital solitary functioning kidneys by measuring renal papilla number. Am J Kidney Dis 72:465–467
Layton AT, Edwards A, Vallon V (2017) Adaptive changes in GFR, tubular morphology, and transport in subtotal nephrectomized kidneys: modeling and analysis. Am J Physiol Renal Physiol 313:F199–F209
Fong D, Denton KM, Moritz KM, Evans R, Singh RR (2014) Compensatory responses to nephron deficiency: adaptive or maladaptive? Nephrology (Carlton) 19:119–128
Lankadeva YR, Singh RR, Tare M, Moritz KM, Denton KM (2014) Loss of a kidney during fetal life: long-term consequences and lessons learned. Am J Physiol Renal Physiol 306:F791-800
Groen In ’t Woud S, Westland R, Feitz WFJ, Roeleveld N, van Wijk JAE, van der Zanden LFM, Schreuder MF (2021) Clinical management of children with a congenital solitary functioning kidney: overview and recommendations. Eur Urol Open Sci 25:11–20
Groen In ’t Woud S, Gobino A, Roeleveld N, van den Heuvel L, Feitz WFJ, van der Zanden LFM, Schreuder MF (2022) Kidney injury rates after unilateral nephrectomy in childhood - a systematic review and meta-analysis. Nephrol Dial Transplant 37:2457–2473
Blantz RC, Pelayo JC (1984) A functional role for the tubuloglomerular feedback mechanism. Kidney Int 25:739–746
Schreuder M, Delemarre-van de Waal H, van Wijk A (2006) Consequences of intrauterine growth restriction for the kidney. Kidney Blood Press Res 29:108–125
Luyckx VA, Brenner BM (2010) The clinical importance of nephron mass. J Am Soc Nephrol 21:898–910
Luyckx VA, Brenner BM (2005) Low birth weight, nephron number, and kidney disease. Kidney Int Suppl:S68–77
Chapman AB (2012) The fetal environment: a critical phase that determines future renal outcomes in autosomal dominant polycystic kidney disease. Kidney Int 81:814–815
Hodgin JB, Rasoulpour M, Markowitz GS, D’Agati VD (2009) Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 4:71–76
Kaze FF, Nguefack S, Asong CM, Assob JCN, Nansseu JR, Kowo MP, Nzana V, Kalla GCM, Halle MP (2020) Birth weight and renal markers in children aged 5–10 years in Cameroon: a cross-sectional study. BMC Nephrol 21:464
Reyes L, Manalich R (2005) Long-term consequences of low birth weight. Kidney Int 68:S107-111
Hostetter TH (2003) Hyperfiltration and glomerulosclerosis. Semin Nephrol 23:194–199
Brenner BM, Lawler EV, Mackenzie HS (1996) The hyperfiltration theory: a paradigm shift in nephrology. Kidney Int 49:1774–1777
Hibino S, Abe Y, Watanabe S, Yamaguchi Y, Nakano Y, Tatsuno M, Itabashi K (2015) Proteinuria caused by glomerular hypertension during adolescence associated with extremely premature birth: a report of two cases. Pediatr Nephrol 30:1889–1892
Kasztan M, Fox BM, Lebensburger JD, Hyndman KA, Speed JS, Pollock JS, Pollock DM (2019) Hyperfiltration predicts long-term renal outcomes in humanized sickle cell mice. Blood Adv 3:1460–1475
Kasztan M, Pollock DM (2019) Impact of ET-1 and sex in glomerular hyperfiltration in humanized sickle cell mice. Clin Sci (Lond) 133:1475–1486
Derebail VK, Zhou Q, Ciccone EJ, Cai J, Ataga KI (2021) Longitudinal study of glomerular hyperfiltration and normalization of estimated glomerular filtration in adults with sickle cell disease. Br J Haematol 195:123–132
Kalk P, Thone-Reineke C, Schwarz A, Godes M, Bauer C, Pfab T, Hocher B (2009) Renal phenotype of ET-1 transgenic mice is modulated by androgens. Eur J Med Res 14:55–58
Polderman KH, Stehouwer CD, van Kamp GJ, Dekker GA, Verheugt FW, Gooren LJ (1993) Influence of sex hormones on plasma endothelin levels. Ann Intern Med 118:429–432
Dousdampanis P, Trigka K, Fourtounas C, Bargman JM (2014) Role of testosterone in the pathogenesis, progression, prognosis and comorbidity of men with chronic kidney disease. Ther Apher Dial 18:220–230
Skrtic M, Lytvyn Y, Bjornstad P, Reich HN, Scholey JW, Yip P, Sochett EB, Perkins B, Cherney DZ (2017) Influence of sex on hyperfiltration in patients with uncomplicated type 1 diabetes. Am J Physiol Renal Physiol 312:F599–F606
Bjornstad P, Cherney DZ (2018) Renal hyperfiltration in adolescents with type 2 diabetes: physiology, sex differences, and implications for diabetic kidney disease. Curr Diab Rep 18:22
Delgadillo D, Barbier O, Sierra G, Reyes JL (2014) Retinoic acid improves recovery after nephrectomy and decreases renal TGF-beta1 expression Gender-related effects. Fundam Clin Pharmacol 28:170–179
Mulroney SE, Woda C, Johnson M, Pesce C (1999) Gender differences in renal growth and function after uninephrectomy in adult rats. Kidney Int 56:944–953
Shapiro JI, Dial LD (2012) How safe is unilateral nephrectomy? Hypertension 60:1383–1384
Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR (2010) Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329:841–845
Ekulu PM, Nkoy AB, Betukumesu DK, Aloni MN, Makulo JRR, Sumaili EK, Mafuta EM, Elmonem MA, Arcolino FO, Kitetele FN, Lepira FB, van den Heuvel LP, Levtchenko EN (2019) APOL1 risk genotypes are associated with early kidney damage in children in sub-Saharan Africa. Kidney Int Rep 4:930–938
Ekulu PM, Nkoy AB, Adebayo OC, Kazadi OK, Aloni MN, Arcolino FO, Ngiyulu RM, Gini JE, Lepira FB, Van den Heuvel LP, Levtchenko EN (2021) A focus on the association of Apol1 with kidney disease in children. Pediatr Nephrol 36:777–788
Fu Y, Zhu JY, Richman A, Zhang Y, Xie X, Das JR, Li J, Ray PE, Han Z (2017) APOL1-G1 in nephrocytes induces hypertrophy and accelerates cell death. J Am Soc Nephrol 28:1106–1116
Ekulu PM, Adebayo OC, Decuypere JP, Bellucci L, Elmonem MA, Nkoy AB, Mekahli D, Bussolati B, van den Heuvel LP, Arcolino FO, Levtchenko EN (2021) Novel Human Podocyte Cell Model Carrying G2/G2 APOL1 High-Risk Genotype. Cells 10:1914
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA (2015) APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 26:3179–3189
Song P, Zhang Y, Yu J, Zha M, Zhu Y, Rahimi K, Rudan I (2019) Global prevalence of hypertension in children: a systematic review and meta-analysis. JAMA Pediatr 173:1154–1163
Bell CS, Samuel JP, Samuels JA (2019) Prevalence of hypertension in children. Hypertension 73:148–152
Colombatti R, Maschietto N, Varotto E, Grison A, Grazzina N, Meneghello L, Teso S, Carli M, Milanesi O, Sainati L (2010) Pulmonary hypertension in sickle cell disease children under 10 years of age. Br J Haematol 150:601–609
Eppens MC, Craig ME, Cusumano J, Hing S, Chan AK, Howard NJ, Silink M, Donaghue KC (2006) Prevalence of diabetes complications in adolescents with type 2 compared with type 1 diabetes. Diabetes Care 29:1300–1306
Davidovits M, Cleper R, Eizenberg N, Hocherman O, Mashiach R (2017) Outcomes of prenatally diagnosed solitary functioning kidney during early life. J Perinatol 37:1325–1329
Schmieder RE, Veelken R, Gatzka CD, Ruddel H, Schachinger H (1995) Predictors for hypertensive nephropathy: results of a 6-year follow-up study in essential hypertension. J Hypertens 13:357–365
Schmieder RE, Messerli FH, Garavaglia G, Nunez B (1990) Glomerular hyperfiltration indicates early target organ damage in essential hypertension. JAMA 264:2775–2780
Larsson L, Aperia A, Wilton P (1980) Effect of normal development on compensatory renal growth. Kidney Int 18:29–35
Krohn AG, Ogden DA, Holmes JH (1966) Renal function in 29 healthy adults before and after nephrectomy. JAMA 196:322–324
Fesler P, Mourad G, du Cailar G, Ribstein J, Mimran A (2015) Arterial stiffness: an independent determinant of adaptive glomerular hyperfiltration after kidney donation. Am J Physiol Renal Physiol 308:F567-571
Abou Jaoude P, Dubourg L, Bacchetta J, Berthiller J, Ranchin B, Cochat P (2011) Congenital versus acquired solitary kidney: is the difference relevant? Nephrol Dial Transplant 26:2188–2194
Donate-Correa J, Ferri CM, Sanchez-Quintana F, Perez-Castro A, Gonzalez-Luis A, Martin-Nunez E, Mora-Fernandez C, Navarro-Gonzalez JF (2021) Inflammatory cytokines in diabetic kidney disease: pathophysiologic and therapeutic implications. Front Med (Lausanne) 7:628289
DiPetrillo K, Gesek FA (2004) Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol 24:352–359
DiPetrillo K, Coutermarsh B, Gesek FA (2003) Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol 284:F113-121
Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, Johnston CI (1998) Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 98:2621–2628
Wang H, Li J, Gai Z, Kullak-Ublick GA, Liu Z (2017) TNF-alpha deficiency prevents renal inflammation and oxidative stress in obese mice. Kidney Blood Press Res 42:416–427
Correia-Costa L, Sousa T, Morato M, Cosme D, Afonso J, Moura C, Mota C, Areias JC, Guerra A, Schaefer F, Caldas Afonso A, Barros H, Albino-Teixeira A, Azevedo A (2016) Association of myeloperoxidase levels with cardiometabolic factors and renal function in prepubertal children. Eur J Clin Invest 46:50–59
Johnson RJ, Couser WG, Chi EY, Adler S, Klebanoff SJ (1987) New mechanism for glomerular injury. Myeloperoxidase-hydrogen peroxide-halide system. J Clin Invest 79:1379–1387
Odobasic D, Kitching AR, Semple TJ, Holdsworth SR (2007) Endogenous myeloperoxidase promotes neutrophil-mediated renal injury, but attenuates T cell immunity inducing crescentic glomerulonephritis. J Am Soc Nephrol 18:760–770
Wang M, Wang Z, Chen Y, Dong Y (2022) Kidney damage caused by obesity and its feasible treatment drugs. Int J Mol Sci 23:747
Briffa JF, McAinch AJ, Poronnik P, Hryciw DH (2013) Adipokines as a link between obesity and chronic kidney disease. Am J Physiol Renal Physiol 305:F1629-1636
Sharma M, Sharma R, McCarthy ET, Savin VJ, Srivastava T (2017) Hyperfiltration-associated biomechanical forces in glomerular injury and response: potential role for eicosanoids. Prostaglandins Other Lipid Mediat 132:59–68
Srivastava T, Hariharan S, Alon US, McCarthy ET, Sharma R, El-Meanawy A, Savin VJ, Sharma M (2018) Hyperfiltration-mediated injury in the remaining kidney of a transplant donor. Transplantation 102:1624–1635
Chagnac A, Zingerman B, Rozen-Zvi B, Herman-Edelstein M (2019) Consequences of glomerular hyperfiltration: the role of physical forces in the pathogenesis of chronic kidney disease in diabetes and obesity. Nephron 143:38–42
Minakawa A, Fukuda A, Sato Y, Kikuchi M, Kitamura K, Wiggins RC, Fujimoto S (2019) Podocyte hypertrophic stress and detachment precedes hyperglycemia or albuminuria in a rat model of obesity and type2 diabetes-associated nephropathy. Sci Rep 9:18485
Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, Wickman LT, Wiggins JE, Muchayi T, Fingar D, Shedden KA, Inoki K, Wiggins RC (2012) Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol 23:1351–1363
Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG (2009) Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 52:691–697
Low S, Zhang X, Wang J, Yeoh LY, Liu YL, Ang KKL, Tang WE, Kwan PY, Tavintharan S, Sum CF, Lim SC (2018) Long-term prospective observation suggests that glomerular hyperfiltration is associated with rapid decline in renal filtration function: a multiethnic study. Diab Vasc Dis Res 15:417–423
Molitch ME, Gao X, Bebu I, de Boer IH, Lachin J, Paterson A, Perkins B, Saenger AK, Steffes M, Zinman B, Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Research Group (2019) Early glomerular hyperfiltration and long-term kidney outcomes in type 1 diabetes: the DCCT/EDIC experience. Clin J Am Soc Nephrol 14:854–861
Acknowledgements
The authors acknowledge Enrico Skoruppa for his assistance in preparing the figures. The figures were created using Biorender.com and Inkscape.
Funding
O.C.A. is supported by the fundamental research grant of Fonds Wetenschappelijk Onderzoek-Vlaanderen (FWO) [grant number 11A5621N]. A.B.N is supported by the Global Minds Scholarship of VLIR-UOS and DGD (Belgium partner in development).
Author information
Authors and Affiliations
Contributions
O.C.A., A.B.N., and L.P.H. searched the literature and wrote the manuscript. O.C.A., A.B.N., L.P.H., V.L., E.L., P.D., and H.P. contributed to the manuscript revision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Answers
1. b; 2. c; 3. c; 4. b; 5. e
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Adebayo, O.C., Nkoy, A.B., van den Heuvel, L.P. et al. Glomerular hyperfiltration: part 2—clinical significance in children. Pediatr Nephrol 38, 2529–2547 (2023). https://doi.org/10.1007/s00467-022-05826-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-022-05826-5