
Abstract
The purpose of this review is to describe Streptococcus pneumoniae–associated hemolytic uremic syndrome (P-HUS) with emphasis on new insights into the pathophysiology and management over the past 10 years. Even though awareness of this clinico-pathological entity has increased, it likely remains under-recognized. Recent observations indicate that although neuraminidase activity and exposure of the T-antigen are necessary for development of P-HUS, they are not sufficient; activation of the alternate pathway of complement may also contribute. It is unclear, however, whether or not eculizumab and/or plasmapheresis are of value.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemolytic uremic syndrome is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury (AKI). It is most commonly caused by Shiga-like toxin-producing Escherichia coli (STEC) but is also a complication of invasive pneumococcal disease (IPD). Atypical forms of hemolytic uremic syndrome (aHUS) include HUS caused by genetic defects resulting in dysregulation of the alternative complement pathway, inborn errors of metabolism, and HUS linked to other neuraminidase-producing organisms [1, 2]. As more is discovered about potential mechanisms for pathophysiology, new categorization schemes for HUS have been proposed [3, 4]. Fakhouri et al. [3] proposed a classification scheme which differentiates HUS with coexisting diseases or conditions, infection-induced HUS, cobalamin C defect-HUS, and atypical HUS (DGKE-HUS, HUS with dysregulation of the complement alternative pathway, HUS without identified cause).
The prevalence of Streptococcus pneumoniae–associated hemolytic uremic syndrome (P-HUS) is highest among children under 2 years of age and is most frequently seen in patients with pneumonia with empyema or meningitis. Waters et al. reported an incidence of empyema of 60% and meningitis of 30% in a cohort with P-HUS [5]. Features of P-HUS usually develop 3 to 13 days (most at 7 to 9 days) after the onset of pneumococcal-related symptoms [6]. In comparison to patients with STEC HUS, those with P-HUS have longer periods of oligo-anuria, more frequent need for acute dialysis, longer hospital stays, a longer duration of thrombocytopenia, and more red blood cell and platelet transfusions [5, 7, 8]. Acute mortality is highest in patients with S. pneumoniae meningitis complicated by HUS [5, 7, 8].
Historical perspective—summary of knowledge prior to 2008
At the time of our previous review in 2008, P-HUS represented about 5% of all reported cases of HUS cases in children and about 40% of HUS cases not caused by STEC [9]. HUS complicating IPD has been recognized since the 1970s and early reports indicated that this rare condition was associated with a very poor clinical outcome. Of the 12 patients reported in the English language literature between 1977 and 1987, 50% died and 67% of the survivors developed chronic kidney disease (CKD) or hypertension. In the following years, the morbidity and mortality rates declined dramatically, likely secondary to advances in critical care management. In reviewing the literature from 1987 to 2007, 73 patients were reported, of whom 12.3% died in the acute phase, 10.1% developed end-stage kidney disease (ESKD), and 16% developed CKD or hypertension [9]. Although 28% had meningitis, 88% of the deaths occurred in this subgroup. In marked contrast, P-HUS not associated with meningitis had a reported acute mortality rate of 2%. The poor prognosis of patients with pneumococcal meningitis complicated by HUS was potentially explained by the earlier observation that the severity of pneumococcal meningitis is related to the neuraminidase activity and Thomsen-Friedenreich antigen (T-antigen) exposure in the central nervous system [10]. Reports of hepatic involvement in the form of hyperbilirubinemia and cholestasis were exceptional at the time of our last report [11, 12], but cases are being increasingly recognized [13, 14]. The mechanism of hepatocellular injury is likely multifactorial, but none of the reported cases had synthetic liver dysfunction, all of the patients had resolution of liver complications, and hepatocellular injury was not associated with increased mortality. Recurrence of P-HUS has not been described, which suggests that, like STEC HUS, there is a single insult to the kidney. Long-term renal outcomes for P-HUS were thought to be analogous to STEC HUS, in which 25–30% of survivors have chronic renal sequelae. The greatest risk factor for the development of CKD after P-HUS was related to the severity of the initial renal injury and need for acute dialysis for greater than 20 days [7].
The lack of a specific laboratory test or universally accepted diagnostic criteria, as well as some overlapping features with disseminated intravascular coagulation (DIC), all likely contributes to cases of P-HUS being under recognized. In our previously proposed P-HUS case definitions, we included definite, probable, and possible cases to aid in recognition and diagnosis of this entity in critically ill patients (Table 1) [7]. HUS and DIC may be difficult to differentiate because both may have microangiopathic hemolytic anemia, thrombocytopenia, and AKI. Bleeding is the predominant clinical manifestation of DIC, but the definition is based on laboratory studies that show significant prolongation of the PT and PTT and consumption of fibrinogen and platelets. Previous classification schemes relied on culture-proven S. pneumoniae infection or histopathologic evidence of thrombotic microangiopathy (TMA). However, the estimated yield of positive blood cultures from patients with pneumonia is only 10–30% [15]. Furthermore, the instability of many of these patients precludes a renal biopsy, and TMA on biopsy is not pathognomonic for P-HUS as it can also occur in DIC. Our criteria for classifying cases of P-HUS incorporated the use of a positive Coombs test in the setting of proven or possible pneumococcal infection as strong evidence for T-antigen activation and, thus, pneumococcal neuraminidase activity. As this laboratory finding is not a feature of DIC or other forms of HUS, its inclusion in the case definition allowed for inclusion of critically ill patients in whom HUS and DIC might coexist.
All serotypes of S. pneumoniae have neuraminidase activity capable of exposing the T-antigen, but different subtypes could theoretically produce varying amounts and activities of neuraminidase that could influence the likelihood of developing HUS. In 2000, a 7-valent (serotypes 4, 6B, 9V, 14, 18C, 19F, 23F) pneumococcal protein conjugate vaccine (PCV7) was licensed for use in young children in the USA and recommended for all children younger than 2 years. Before 2007, the serotype most frequently associated with the development of P-HUS was serotype 14 as it was detected in 35% of the 23 reported cases [7]. Several years after the introduction of the PCV7 vaccine, the rate of serotype 19A invasive pneumococcal disease in children younger than 5 years increased significantly. In 2007, Waters et al. compiled the largest series of bacteriologic data from 12 patients with pneumococcal HUS in the UK [5]. Eleven of the pneumococcal isolates were from the post-pneumococcal vaccine era: 6 (54.5%) were serotype 19A, 2 were serotype 14, 2 were serotype 3, and 1 was serotype 6A. In a case series of 14 patients diagnosed with P-HUS at The Children’s Hospital of Philadelphia between 1988 and 2009, there was serotype data for 12 patients: 8 (66.7%) had serotype 19A, 2 had serotype 14, 1 had serotype 9 V, and 1 had serotype 6A. In this cohort, all 6 confirmed infections after the induction of the 7-valent pneumococcal protein conjugate vaccine was caused by serotype 19A [7].
The precise pathophysiology of P-HUS had not been determined. However, there has been evidence for a role for neuraminidase activity and exposure of the T-antigen, which is normally hidden by neuraminic acid. Klein et al. speculated that S. pneumoniae neuraminidases are released in the circulation and remove sialic acid (N-acetylneuraminic acid) from the cell surface glycans of red blood cells, platelets, and glomerular endothelial cells leading to the exposure of the T-antigen to which preformed host IgM antibodies bind [16]. It was believed that this antibody binding initiated the cascade of events that led to P-HUS. Thus, it was previously accepted that clinicians should be judicious in the transfusion of blood products and only use washed products in order to prevent further exposure to preformed antibody [6, 17]. While this strategy is still widely in use, new studies investigating the underlying molecular pathology of P-HUS question the necessity of this approach and suggest other strategies for limiting thrombotic microangiopathy, hemolysis, and renal injury.
New insights into pathophysiology
Over the past 10 years, even with improved vaccine coverage and a reduction in the prevalence of IPD, pneumococcal infection remains a major cause of HUS in children. While STEC infection remains the leading cause of HUS, the incidence of P-HUS appears to be increasing [18,19,20]. P-HUS now represents approximately 5–15% of all reported HUS cases. This may be due to increased recognition or possibly a shift in serotype of IPD associated with increased vaccination rates or serotype coverage worldwide. Prior to the era of pneumococcal vaccination, serotypes most likely to cause HUS were 3, 6B, 8, 9 V, 14, 19, and 23F [6, 20]. After the introduction of PCV7 in 2000, the emergence of non-vaccine serotypes 19A, 1, 3, 6A, and 7F was observed [5,6,7,8, 19, 20]. In 2010, PCV13 was introduced which includes protection against serotype 19A. One group in the UK studying IPD rates in infant population recently found that the majority of infections (71.4%) in the post-PCV13 era in which serotype data were available were caused by non-PCV 13 serotypes [21]. Based on currently published data, pneumococcal serotype 19A may still remain the most commonly reported serotype associated with P-HUS despite the introduction of the PCV13 vaccine [20]. However, there are limited available epidemiological serotype data after introduction of universal 13-valent vaccine introduction in patients who develop P-HUS.
Despite the severity of P-HUS and an increase in awareness and diagnosis, the pathogenesis is still not definitively understood, and clinicians cannot accurately predict which patients with IPD will develop this complication. As previously mentioned, it has long been thought that pneumococcal neuraminidase contributes in some way to the development of P-HUS [16]. However, its exact role and the downstream effects of the desialylation of glycoproteins on the surface of red blood cells (RBCs) and glomerular endothelial cells are not fully understood. Neuraminidase released by S. pneumoniae acts by cleaving terminal N-acetylneuraminic acid (sialic acid) from glycoproteins present in plasma and on the cell surfaces in contact with the enzyme (red blood cells, platelets, glomerular endothelial cells). This exposes the cryptic T-antigen. This process is known as T-activation. In 2008, the best evidence for the role of neuraminidase in the pathogenesis of P-HUS was supported by the study by Huang and colleagues who showed that T-activation was significantly associated with HUS in IPD. Using the peanut (Arachis hypogaea) lectin agglutinin assay, they demonstrated T-antigen exposure in 100% (13/13) of children with P-HUS, 67% (6/9) patients with IPD associated hemolytic anemia, and only 43% (6/14) children with uncomplicated IPD [22]. Thus, T-antigen exposure was 100% sensitive, but only 48% specific for P-HUS in IPD. Other studies have subsequently confirmed 100% sensitivity of the peanut lectin agglutination assay for P-HUS [23]. This assay is also specific for pneumococcal infection as compared to healthy controls [24]. Nevertheless, because only 0.4–0.6% of IPD patients progress to P-HUS, additional factors must be involved [6, 9].
The notion that T-antigen exposure and subsequent binding of preformed IgM antibodies led to the development of HUS was supported by the detection of these erythrocyte-bound antibodies by the direct Coombs test. This test was initially reported to be positive in about 90% of cases of P-HUS [7, 25]. More recent data suggests that the sensitivity of Coombs positivity is closer to 60% [24, 26]. Doubt has been cast on this theory as anti-T antibodies are low-titer antibodies with extremely weak hemolyzing capacity and have never been definitely shown to cause hemolysis in patients with T-activated RBCs [27,28,29]. Furthermore, it is a cold reactive antibody and at 37 °C causes neither red blood cell agglutination nor complement activation [28]. Finally, P-HUS may occur in children as young as 4 months of age when a significant amount of anti-T is unlikely to be present [24]. Thus, it is entirely possible that there may be other downstream effects of neuraminidase activity and T-antigen exposure that lead to P-HUS separately from IgM binding.
As the shift away from the central role of IgM binding to the T-antigen in the pathogenesis of P-HUS has gained wider acceptance, several studies have attempted to better define the role of neuraminidase activity. Samples collected during the acute phase of illness from a 9-month-old patient with P-HUS were analyzed with mass spectrometry. Decreased sialylation was found in both transferrin N-glycans and IgA1 O-glycans as compared to three healthy controls [30, 31]. The authors concluded that the decreased sialylation was a result of increased serum neuraminidase activity from enzymes secreted by the pneumococcal bacteria in the affected infant. A separate study of 10 patients with IPD (5 with HUS, 3 with hemolytic anemia alone, and 2 patients with neither) hinted at the central role of neuraminidase activity and sensitivity of T-antigen testing for the early diagnosis of P-HUS [24]. Red blood cell T-antigen testing with both peanut lectin and Glycine soja lectin was 100% sensitive for P-HUS, but using both lectin assays together or quantifying T-antigen expression improved the specificity of diagnosis. Red blood cells from the two patients with uncomplicated IPD reacted with peanut lectin, but not Glycine soja. Additionally, the agglutination titer was much lower for those patients without hemolytic anemia or HUS. In contrast, the Coombs test was positive only in 3 of 5 patients with HUS and 2 of 3 with hemolytic anemia, further suggesting that neuraminidase activity/T-antigen exposure is the crucial step rather than native immunoglobulin binding. However, while the peanut and Glycine soja assays seem to improve the sensitivity and specificity of diagnosing P-HUS, they are not readily available in most laboratories, thereby reducing clinical utility. While there are no data regarding the rate of Coombs positivity in pneumococcal infection without HUS, in this small study, neither of the patients with uncomplicated IPD had a positive Coombs test. In light of these facts, in clinical practice, a positive direct Coombs test in a child with HUS and signs of infection remains an indicator of P-HUS.
S. pneumoniae expresses up to three types of sialidases (NanA, NanB, and NanC). NanA is invariably present in pneumococcal strains isolated from clinical samples of patients with IPD and exposure of T-antigen in vivo is dependent on NanA [27,28,29]. However, Smith et al. attempted to determine the importance of NanA and its genomic diversity in the pathogenesis in P-HUS. They found significant difference neither in overall neuraminidase activity in vitro nor in genetic content between the isolates from patients with P-HUS compared to control isolates from patients with IPD without HUS [32]. The lack of an association between different NanA alleles in P-HUS isolates compared with controls suggests that host factors play a more significant role in the development of HUS compared to any specific NanA variant. In contrast, another group examined the distribution of the three neuraminidase genes in pneumococcal isolates from 18 children with P-HUS and 54 controls with IPD without HUS [23]. They found that all isolates from both groups had genes NanA and NanB and also identified a significantly higher carriage rate of NanC in the causative pneumococcal isolates from patients with P-HUS (89% vs 41%). The sensitivity and specificity of the presence of NanC in predicting the development of P-HUS in this population were 89% and 59% respectively [23]. The result suggests that NanC could provide an additive effect to NanA and NanB in the overall activity of pneumococcal neuraminidases in exposing T-antigen on various cells in patients with HUS. This finding was not replicated in a subsequent study which compared 29 isolates from patients with P-HUS and serotype-matched controls with IPD [33]. In this study, no difference was observed in NanC distribution and there was no significant correlation between neuraminidase activity and disease state. It is also possible that NanC may be important for tissue-specific virulence as one study demonstrated that the prevalence of NanC activity was 1.4 times higher among samples taken from the CSF of children with IPD as compared to isolates taken from the upper respiratory tract of carriers [34]. Importantly, these isolates were not taken from patients with HUS but could be relevant to predicting clinical course as mortality in P-HUS is highest in those with meningitis. Overall, there have been conflicting reports regarding whether the degree of neuraminidase activity and antigen exposure correlates with development or severity of HUS, with some studies reporting very high neuraminidase activity in P-HUS [24, 35] and others not substantiating this finding [32, 33]. It is clear that neuraminidase activity and T-activation are associated with S. pneumoniae infection and are likely involved in the pathogenesis of P-HUS, but T-antigen exposure alone does not seem to be sufficient in causing P-HUS. It remains unclear whether microbial factors, host factors, or a combination of both contributes to the development of P-HUS.
Nicolas Burin des Roziers et al. have hypothesized that galectin-3, the major endogenous T-antigen-binding protein, may play a role in P-HUS pathogenesis [24]. Gal-3 is expressed and secreted by macrophages and other immune cells and is implicated in the inflammatory response and host defense against pneumococcal infection [24, 36]. Additionally, Gal-3 oligomerizes at high concentrations and forms heterogeneous multimolecular complexes that cross-link both cell surfaces and glycoconjugates expressing the T-antigen [37]. They found that plasma concentrations of Gal-3 were substantially higher in all 10 patients with IPD as compared to 29 healthy controls [24]. The average plasma concentration of healthy controls was 1.2 ± 0.6 ng/mL, while patients with IPD without HUS had levels between 8 and 26 ng/mL and those with P-HUS had levels ranging from 22 to 41 ng/mL. The authors suggest that the high concentrations of Gal-3 protein in P-HUS patients may contribute increased adhesion between desialylated RBCs and endothelial cells both expressing T-antigen, which could in turn generate complement activation in close contact with the endothelial cell and ultimately HUS [24].
Another hypothesis that speaks to the potential contribution of a host factor is the observation that the transient dysregulation of complement activation may lead to the development of TMA and P-HUS. In addition to unmasking T-antigen, desialylation on red blood cells also triggers complement activation via the alternative pathway in vitro and in vivo in guinea pigs [38, 39]. This may be due to decreased regulation of the pathway because factor H relies on membrane sialic acid residues for binding. As a result of neuraminidase desialylation, factor H cannot bind properly to C3 convertase on the cell surface, leading to complement activation and cellular injury [40, 41]. Kerr et al. demonstrated that the complex of CFH, C3b, and glycosaminoglycan/sialic acid suppresses complement activation more effectively than the CFH-C3b complex formed on surfaces lacking glycosaminoglycan and sialic acids [41]. Furthermore, this may play a larger role in determining which patients with IPD will ultimately develop HUS and may lead to overlap in diagnosis and treatment strategies with atypical forms of HUS (aHUS) caused by defects in the regulation of the alternative complement pathway. One case series where genetic testing was performed on five patients with P-HUS identified mutations in three patients in genes previously associated with aHUS: one published CFI variant and two novel mutations in the CFH and THBD genes [1]. Additionally, several studies have found that the concentrations of C3 and C4 and the activity of the classical and alternative pathways were decreased in patients with P-HUS, indicating severe activation and complement consumption [1, 42, 43]. Therefore, it is possible that individuals with genetic variations in complement pathway regulation who experience IPD and the subsequent loss of terminal sialic acid residues from host glycans are most likely to develop P-HUS, but there is limited data to support these claims at this time.
Meinel et al., in contrast, proposed a mechanism for disruption of the endothelial cell layer and creation of a local thrombogenic state that may result in P-HUS which is entirely independent of neuraminidase activity and host genes [44]. HUS-inducing pneumococci isolated from two patients during the acute phase of illness were shown to bind human plasminogen efficiently via a rare variant of a bacterial surface protein PspC. The plasminogen attached to these PspC variants was converted to plasmin at the bacterial surface and the active protease degraded fibrinogen and cleaved C3b disrupting local complement homeostasis and thereby causing damage to endothelial cells [44].
Management
Treatment strategies still consist of supportive care measures: appropriate antimicrobial therapy, fluid and blood product resuscitation, and renal replacement therapy. Current research suggests that it is unlikely that anti-T IgM binding is a critical step in the molecular pathology of P-HUS. Its binding, when detected, is likely associated with P-HUS as a marker of neuraminidase activity and desialylation of membrane glycoproteins rather than causative of downstream disease pathology. Thus, washing of packed RBCs and avoidance of fresh frozen plasma are likely not necessary to prevent worsening of the thrombotic microangiopathy. Alternatively, the focus on appropriate therapies is likely to shift towards the potential roles of either inhibition of neuraminidase activity or complement blockade.
Most humans develop neuraminidase-neutralizing antibodies during the first 2 years of life [42, 45,46,47]. Decreased antibody development may be part of the reason that younger patients are more likely to develop P-HUS in the setting of IPD. It has been suggested that intravenous immune globulin (IVIG) could be used to inactivate circulating pneumococcal neuraminidase [42]. Data from one patient treated with high-dose IVIG demonstrated that hemolysis and thrombocytopenia steadily improved after each dose of IVIG [42]. Plasmapheresis could theoretically remove neuraminidase, but there is insufficient evidence to support its use in P-HUS.
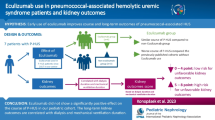
Given the increasing interest in a role of unregulated complement activity in the development of P-HUS and the availability of a terminal complement inhibitor eculizumab, the question of its use in this disease process is inevitable. There are two reported cases of patients with definite P-HUS who were treated with eculizumab [43, 48]. A 21-month-old female had a low C3 during the acute phase of illness that normalized just prior to eculizumab administration. After her first dose of the medication between days 7 and 10 of illness, there were an immediate increase in platelets, a reported improvement in irritability and slow improvement in renal function. There were no detectable abnormalities in the regulation of the alternative complement pathway. Eculizumab was stopped after four doses. A 53-year-old man with PCR-confirmed pneumococcal infection, microangiopathic hemolytic anemia, and acute kidney injury who was unresponsive to plasma exchange was treated with eculizumab. Three days after the first injection, his platelet count began to improve. He began to make urine again on day 10, and dialysis was stopped on day 14. Genetic screening showed no variants of any complement genes. Eculizumab therapy was maintained for 6 months, and he had sustained normalization of all biological parameters [48]. The benefit of short-term complement inhibition and suppression of C5a formation is unclear at this time, and as with plasmapheresis, there is insufficient evidence to recommend this treatment empirically. However, a conservative approach may be to use eculizumab in severe cases of P-HUS who have reduced C3 concentrations. Furthermore, genetic screening for complement variants in patients with a diagnosis of P-HUS may help to shed light on the potential role of dysregulation in this pathway in the development of P-HUS and have implications in management decisions.
Conclusion
Despite improved vaccine coverage and a reduction in the prevalence of IPD, pneumococcal infection remains a major cause of HUS in children. The understanding of the molecular pathology of this disease process is improving, but advances in clinical diagnosis and management remain limited. It is clear that pneumococcal neuraminidase activity plays a central role in the pathogenesis of P-HUS and T-activation is a sensitive marker of its activity. The specificity of T-antigen testing for P-HUS can be improved by using a combination of peanut and Glycine soja lectin assays or by quantifying T-antigen expression. However, these results have not been replicated and the clinical availability of these assays is limited. Several studies support a role for excessive complement activation in the acute phase of illness, whether via decreased Factor H binding to desialylated membranes or PspC binding plasminogen with activation to plasmin causing endothelial injury. Pathogenic variants of genes involved in the regulation of the alternative complement pathway may predispose affected individuals to P-HUS via excessive complement activation in the setting of IPD; however, there is limited data available on the number of patients who have developed P-HUS with these types of mutations. Detailed clinical and experimental investigations are warranted to better understand the role of unregulated complement activation in P-HUS and whether its transient blockade with eculizumab during the acute phase of illness would improve outcomes.
References
Szilágyi A, Kiss N, Bereczki C, Tálosi G, Rácz K, Túri S, Györke Z, Simon E, Horváth E, Kelen K, Reusz GS, Szabó AJ, Tulassay T, Prohászka Z (2013) The role of complement in Streptococcus pneumoniae-associated haemolytic uraemic syndrome. Nephrol Dial Transplant 28:2237–2245. https://doi.org/10.1093/ndt/gft198
Kaplan BS, Ruebner RL, Spinale JM, Copelovitch L (2014) Current treatment of atypical hemolytic uremic syndrome. Intractable Rare Dis Res 3:34–45. https://doi.org/10.5582/irdr.2014.01001
Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet 390:681–696. https://doi.org/10.1016/S0140-6736(17)30062-4
Ruebner RL, Kaplan BS, Copelovitch L (2012) A time for reappraisal of “atypical” hemolytic uremic syndrome: should all patients be treated the same? Eur J Pediatr 171:1519–1525. https://doi.org/10.1007/s00431-012-1763-z
Waters AM, Kerecuk L, Luk D, Haq MR, Fitzpatrick MM, Gilbert RD, Inward C, Jones C, Pichon B, Reid C, Slack MPE, Van’t Hoff W, Dillon MJ, Taylor CM, Tullus K (2007) Hemolytic uremic syndrome associated with invasive pneumococcal disease: the United Kingdom experience. J Pediatr 151:140–144. https://doi.org/10.1016/j.jpeds.2007.03.055
Spinale JM, Ruebner RL, Kaplan BS, Copelovitch L (2013) Update on Streptococcus pneumoniae associated hemolytic uremic syndrome. Curr Opin Pediatr 25:203–208. https://doi.org/10.1097/MOP.0b013e32835d7f2c
Copelovitch L, Kaplan BS (2010) Streptococcus pneumoniae-associated hemolytic uremic syndrome: classification and the emergence of serotype 19A. Pediatrics 125:e174–e182. https://doi.org/10.1542/peds.2007-2017
Banerjee R, Hersh AL, Newland J, Beekmann SE, Polgreen PM, Bender J, Shaw J, Copelovitch L, Kaplan BS, Shah SS, Emerging Infections Network Hemolytic-Uremic Syndrome Study Group (2011) Streptococcus pneumoniae-associated hemolytic uremic syndrome among children in North America. Pediatr Infect Dis J 30:736–739. https://doi.org/10.1097/INF.0b013e3182191c58
Copelovitch L, Kaplan BS (2008) Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol 23:1951–1956. https://doi.org/10.1007/s00467-007-0518-y
Vierbuchen M, Klein PJ (1983) Histochemical demonstration of neuraminidase effects in pneumococcal meningitis. Lab Investig 48:181–186
Chen J-P, Chen S-M, Sheu J-N (2007) Unusual manifestation of severe conjugated hyperbilirubinemia in an infant with Streptococcus pneumoniae-associated hemolytic uremic syndrome. J Formos Med Assoc 106:S17–S22. https://doi.org/10.1016/S0929-6646(09)60347-8
Pan CG, Leichter HE, Werlin SL (1995) Hepatocellular injury in Streptococcus pneumoniae-associated hemolytic uremic syndrome in children. Pediatr Nephrol 9:690–693
Patel MG, Porto AF (2013) Conjugated hyperbilirubinemia in a child with Streptococcus pneumoniae-associated Hemolytic Uremic Syndrome. ACG Case Rep J 1:64–67. https://doi.org/10.14309/crj.2013.22
Anastaze Stelle K, Cachat F, Perez M-H, Chehade H (2016) Streptococcus pneumoniae–associated hemolytic and uremic syndrome with cholestasis: a case report and brief literature review. Clin Pediatr (Phila) 55:189–191. https://doi.org/10.1177/0009922815580406
Obaro SK, Madhi SA (2006) Bacterial pneumonia vaccines and childhood pneumonia: are we winning, refining, or redefining? Lancet Infect Dis 6:150–161. https://doi.org/10.1016/S1473-3099(06)70411-X
Klein PJ, Bulla M, Newman RA, Müller P, Uhlenbruck G, Schaefer HE, Krüger G, Fisher R (1977) Thomsen-Friedenreich antigen in haemolytic-uraemic syndrome. Lancet 2:1024–1025
Oliver JW, Akins RS, Bibens MK, Dunn DM (2010) Pneumococcal induced T-activation with resultant thrombotic microangiopathy. Clin Med Insights Pathol 3:13–17
Veesenmeyer AF, Edmonson MB (2013) Trends in US hospital stays for Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Infect Dis J 32:731–735. https://doi.org/10.1097/INF.0b013e31828b31c8
Bender JM, Ampofo K, Byington CL, Grinsell M, Korgenski K, Daly JA, Mason EO, Pavia AT (2010) Epidemiology of Streptococcus pneumoniae-induced hemolytic uremic syndrome in Utah children. Pediatr Infect Dis J 29:712–716. https://doi.org/10.1097/INF.0b013e3181db03a7
Lawrence J, Gwee A, Quinlan C (2018) Pneumococcal haemolytic uraemic syndrome in the postvaccine era. Arch Dis Child 103:957–961. https://doi.org/10.1136/archdischild-2017-313923
Kent A, Makwana A, Sheppard CL, Collins S, Fry NK, Heath PT, Ramsay M, Ladhani SN (2019) Invasive pneumococcal disease in UK children <1 year of age in the post-13-valent pneumococcal conjugate vaccine era: what are the risks now? Clin Infect Dis 69:84–90. https://doi.org/10.1093/cid/ciy842
Huang DT-N, Chi H, Lee H-C, Chiu N-C, Huang F-Y (2006) T-antigen activation for prediction of pneumococcus-induced hemolytic uremic syndrome and hemolytic anemia. Pediatr Infect Dis J 25:608–610. https://doi.org/10.1097/01.inf.0000223494.83542.ad
Janapatla R-P, Hsu M-H, Hsieh Y-C, Lee H-Y, Lin T-Y, Chiu C-H (2013) Necrotizing pneumonia caused by nanC-carrying serotypes is associated with pneumococcal haemolytic uraemic syndrome in children. Clin Microbiol Infect 19:480–486. https://doi.org/10.1111/j.1469-0691.2012.03894.x
Burin des Roziers N, Chadebech P, Bodivit G, Guinchard E, Bruneel A, Dupré T, Chevret L, Jugie M, Gallon P, Bierling P, Noizat-Pirenne F (2015) Red blood cell Thomsen-Friedenreich antigen expression and galectin-3 plasma concentrations in Streptococcus pneumoniae-associated hemolytic uremic syndrome and hemolytic anemia. Transfusion (Paris) 55:1563–1571. https://doi.org/10.1111/trf.12981
von Vigier RO, Fossali E, Crosazzo L, Bianchetti MG (2005) Positive Coombs test in postpneumococcal hemolytic-uremic syndrome. Pediatr Infect Dis J 24:1028–1029
Loupiac A, Elayan A, Cailliez M, Adra A-L, Decramer S, Thouret M-C, Harambat J, Guigonis V (2013) Diagnosis of Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Infect Dis J 32:1045–1049. https://doi.org/10.1097/INF.0b013e31829ee872
Burin des Roziers N, Bodivit G, Chadebech P, Nzouakou R, Bierling P, Noizat-Pirenne F (2011) Anti-T haemolysins: the effects of sialic acid removal and 2-aminoethylisothiouronium bromide treatment of erythrocytes on immune lysis. Vox Sang 100:401–408. https://doi.org/10.1111/j.1423-0410.2010.01450.x
Crookston KP, Reiner AP, Cooper LJ, Sacher RA, Blajchman MA, Heddle NM (2000) RBC T activation and hemolysis: implications for pediatric transfusion management. Transfusion (Paris) 40:801–812
Eder AF, Manno CS (2001) Does red-cell T activation matter? Br J Haematol 114:25–30
Aoki H, Shiomi M, Ikeda T, Ishii T, Shimizu N, Togawa M, Okamoto N, Kadoya M, Wada Y (2013) Decreased sialylation of IgA1 O-glycans associated with pneumococcal hemolytic uremic syndrome. Pediatr Int 55:e143–e145. https://doi.org/10.1111/ped.12166
Wada Y, Azadi P, Costello CE, Dell A, Dwek RA, Geyer H, Geyer R, Kakehi K, Karlsson NG, Kato K, Kawasaki N, Khoo K-H, Kim S, Kondo A, Lattova E, Mechref Y, Miyoshi E, Nakamura K, Narimatsu H, Novotny MV, Packer NH, Perreault H, Peter-Katalinic J, Pohlentz G, Reinhold VN, Rudd PM, Suzuki A, Taniguchi N (2007) Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 17:411–422. https://doi.org/10.1093/glycob/cwl086
Smith A, Johnston C, Inverarity D, Slack M, Paterson GK, Diggle M, Mitchell T (2013) Investigating the role of pneumococcal neuraminidase a activity in isolates from pneumococcal haemolytic uraemic syndrome. J Med Microbiol 62:1735–1742. https://doi.org/10.1099/jmm.0.063479-0
Singh AK, Osman AS, Woodiga SA, White P, Mahan JD, King SJ (2016) Defining the role of pneumococcal neuraminidases and O-glycosidase in pneumococcal haemolytic uraemic syndrome. J Med Microbiol 65:975–984. https://doi.org/10.1099/jmm.0.000322
Pettigrew MM, Fennie KP, York MP, Daniels J, Ghaffar F (2006) Variation in the presence of neuraminidase genes among Streptococcus pneumoniae isolates with identical sequence types. Infect Immun 74:3360–3365. https://doi.org/10.1128/IAI.01442-05
de Loos F, Huijben KMLC, van der Kar NCAJ, Monnens LAH, van den Heuvel LPWJ, Groener JEM, de Moor RA, Wevers RA (2002) Hemolytic uremic syndrome attributable to Streptococcus pneumoniae infection: a novel cause for secondary protein N-glycan abnormalities. Clin Chem 48:781–784
Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG (2002) Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol 168:1813–1822
Ahmad N, Gabius H-J, André S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF (2004) Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem 279:10841–10847. https://doi.org/10.1074/jbc.M312834200
Fearon DT (1978) Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc Natl Acad Sci U S A 75:1971–1975
Brown EJ, Joiner KA, Frank MM (1983) Interaction of desialated guinea pig erythrocytes with the classical and alternative pathways of guinea pig complement in vivo and in vitro. J Clin Invest 71:1710–1719
Ault BH (2000) Factor H and the pathogenesis of renal diseases. Pediatr Nephrol 14:1045–1053
Kerr H, Wong E, Makou E, Yang Y, Marchbank K, Kavanagh D, Richards A, Herbert AP, Barlow PN (2017) Disease-linked mutations in factor H reveal pivotal role of cofactor activity in self-surface-selective regulation of complement activation. J Biol Chem 292:13345–13360. https://doi.org/10.1074/jbc.M117.795088
Bitzan M, AlKandari O, Whittemore B, Yin X-L (2018) Complement depletion and Coombs positivity in pneumococcal hemolytic uremic syndrome (pnHUS). Case series and plea to revisit an old pathogenetic concept. Int J Med Microbiol 308:1096–1104. https://doi.org/10.1016/j.ijmm.2018.08.007
Gilbert RD, Nagra A, Haq MR (2013) Does dysregulated complement activation contribute to haemolytic uraemic syndrome secondary to Streptococcus pneumoniae? Med Hypotheses 81:400–403. https://doi.org/10.1016/j.mehy.2013.05.030
Meinel C, Spartà G, Dahse H-M, Hörhold F, König R, Westermann M, Coldewey SM, Cseresnyés Z, Figge MT, Hammerschmidt S, Skerka C, Zipfel PF (2018) Streptococcus pneumoniae from patients with hemolytic uremic syndrome binds human plasminogen via the surface protein PspC and uses plasmin to damage human endothelial cells. J Infect Dis 217:358–370. https://doi.org/10.1093/infdis/jix305
Eber SW, Polster H, Quentin SH, Rumpf KW, Lynen R (1993) Hemolytic-uremic syndrome in pneumococcal meningitis and infection. Importance of T-transformation. Monatsschr Kinderheilkd 141:219–222
Seges RA, Kenny A, Bird GW, Wingham J, Baals H, Stauffer UG (1981) Pediatric surgical patients with severe anaerobic infection: report of 16 T-antigen positive cases and possible hazards of blood transfusion. J Pediatr Surg 16:905–910
Simell B, Jaakkola T, Lahdenkari M, Briles D, Hollingshead S, Kilpi TM, Käyhty H (2006) Serum antibodies to pneumococcal neuraminidase NanA in relation to pneumococcal carriage and acute otitis media. Clin Vaccine Immunol 13:1177–1179. https://doi.org/10.1128/CVI.00257-06
Jeantet G, Pernin V, Brunot V, Roccabianca A, Macombe A, Szwarc I, Klouche K, Loirat C, Mourad G, Frémeaux-Bacchi V, Le Quintrec M (2019) Successful treatment of a Streptococcus pneumoniae-associated haemolytic uraemic syndrome by eculizumab. Clin Kidney J 12:106–109. https://doi.org/10.1093/ckj/sfy019
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scobell, R.R., Kaplan, B.S. & Copelovitch, L. New insights into the pathogenesis of Streptococcus pneumoniae–associated hemolytic uremic syndrome. Pediatr Nephrol 35, 1585–1591 (2020). https://doi.org/10.1007/s00467-019-04342-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-019-04342-3