
Abstract
It is indisputable that immunosuppressive therapy and pathological diagnosis of renal biopsy have greatly improved the prognosis of childhood nephrotic syndrome. Unfortunately, there is no “one-size-fits-all” approach for precise patient stratification and treatment when facing the huge challenges posed by steroid-resistant nephrotic syndrome (SRNS). But genomic medicine has brought a glimmer of light, and the cognition of SRNS has entered a new stage. Based on this, identification of single genetic variants of SRNS has recognized the key role of podocyte injury in its pathogenesis. Targeted treatment of podocyte injury is paramount, and immunosuppressant with podocyte-targeted therapy seems to be more suitable as the first choice for SRNS, that is, we need to pay attention to their additional non-immunosuppressive effects. In the same way, other effect factors of nephrotic syndrome and the related causes of immunosuppressive therapy resistance require us to select reasonable and targeted non-immunosuppressive therapies, instead of only blindly using steroids and immunosuppressants, which may be ineffective and bring significant side effects. This article provides a summary of the clinical value of identification of genetic variants in podocytes and non-immunosuppressive therapy for nephrotic syndrome in children.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nephrotic syndrome, a common pediatric kidney disease with idiopathic condition, is characterized by severe proteinuria, hypoproteinemia, and generalized edema. Nephrotic syndrome in children can have a frequently relapsing course complicated with infection, venous thromboembolism, and acute kidney injury. The prognosis of nephrotic syndrome was usually very poor prior to the introduction of corticosteroid and antibiotic therapy, and the mortality rate in children was nearly 67% [1]. However, with the widespread use of immunosuppressive therapy combined with the guidance of pathologic diagnosis by renal biopsy, the mortality rate dramatically reduced to 3% or less [2]. Unfortunately, there is no one-size-fits-all treatment for nephrotic syndrome in children due to the etiological heterogeneity. Although high remission has been achieved in children with idiopathic nephrotic syndrome, approximately 12–15% of children with steroid-resistant nephrotic syndrome (SRNS) undergoing renal biopsy do not respond to immunosuppressive therapy, and 50% of them will progress to end-stage renal disease (ESRD) within 15 years [3]. Up to now, even with great efforts and various new drugs, the evaluation and treatment of nephrotic syndrome is still posing a persistent challenge for clinical practice.
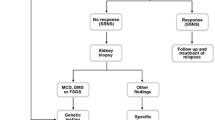
In general, minimal change disease (MCD) is the most common pathological finding in childhood nephrotic syndrome, and corticosteroids will induce remission in more than 90% of children patients with MCD [4]. Thus, childhood nephrotic syndrome can be treated with empirical steroid therapy first, and most patients will be relieved. However, once SRNS occurs in children, renal biopsy is necessary to determine the possible etiology. Generally, focal segmental glomerulosclerosis (FSGS) is the most common pathological type of SRNS in children. Thus, kidney biopsy is usually recommended to obtain a histological diagnosis in children with SRNS, whose pathological type is doubted [5]. Extensive research has proposed the podocyte as a crucial site of cellular injury in FSGS. Since Kestila et al. identified the mutation in NPSH1 related to nephrin as a cause of congenital nephrotic syndrome in 1998 [6], more than 50 genes have been identified as essential for podocyte development, structure and function, which have been proved to play a key role in the pathogenesis of SRNS and/or FSGS [7]. In these children patients, podocyte gene detection will contribute to rational treatment decisions.
Expanding our knowledge of podocyte molecular mechanisms will aid physicians to redefine the physiopathological understanding, diagnostic assessment, and prognostic judgment in children affected with nephrotic syndrome. Moreover, a better understanding of these mechanisms can help the development of specific targeted therapies, and these patients can also be spared ineffective immunosuppressive drug-based treatments that may have marked adverse effects. Therefore, it is time to consider some valuable non-immunosuppressive therapies as the main body or necessary supplement, which will hold promise in the treatment of nephrotic syndrome.
Updating nephrotic syndrome
Over the past two decades, the understanding of nephrotic syndrome has fundamentally transformed with the discovery of ever-increasing genetic disorders of podocytes. These novel insights redefine diagnostic classification and prognostic assessment, as well as clinical routine management in childhood-onset nephrotic syndrome.
Clinical features of nephrotic syndrome in children
Idiopathic nephrotic syndrome is clinically classified based on response to corticosteroid therapy. At the time of first presentation, 80–90% of children patients over 1 year of age achieve complete remission with 4 weeks of steroid treatment (steroid-sensitive nephrotic syndrome (SSNS)) [8, 9], and 60–70% of them have either frequently relapsing nephrotic syndrome (FRNS, ≥ 2 relapses in first 6 months or ≥ relapses in any 1-year period) or steroid-dependent nephrotic syndrome (SDNS, relapse while on steroid therapy or within 2 weeks after steroid cessation) [10,11,12]. According to the current definition for idiopathic nephritic syndrome by the 2012 Kidney Disease Improving Global Outcomes (KDIGO) guidelines, Steroid resistance is defined as the absence of complete remission of proteinuria despite 8 weeks of therapy with prednisone at a dose of 2 mg/kg/day, which is adopted from the definition by the International study of Kidney Disease in Children (ISKDC) [9, 13]. Absence of remission despite therapy with daily prednisolone at a dose of 2 mg/kg/day or 60 mg/m2/day for 4 weeks is also considered as SRNS [14, 15] (Table 1). Steroid resistance usually not only occurs during initial treatment with prednisone (initial resistance) but can also occur because of relapse in patients who had previously responded well to therapy with steroids (late resistance). Eventually, about 20% of patients with nephrotic syndrome are steroid-resistant, including some FRNS patients who are initially steroid sensitive [16]. Patients with SRNS can also show a multidrug-resistant phenotype, which has been proven to bring more challenge to clinical therapy. Of steroid-resistant patients, 36~50% will progress to ESRD within 10 years according to cohort studies [3].
Genetic variants of podocyte injury in nephrotic syndrome
Over the past two decades, plentiful research has highlighted the importance of the podocyte as a key site of cellular injury in nephrotic syndrome [17]. These studies have demonstrated that intracellular proteins and molecular pathways regulating podocyte structure and function play crucial roles in the development of nephrotic syndrome and the response of patients. As mentioned above, the pioneering discovery of genes encoding the slit membrane proteins nephrin (NPHS1) and podocin (NPHS2) has promoted the identification of more than 50 genes expressed in podocytes that are related to the pathogenesis of different subtypes of SRNS. Most of the encoded proteins can be grouped into distinct structural protein complexes and signaling pathways within the podocyte, including those involved in slit diaphragm structure and function (NPHS1, NPHS2, PLCE1, CD2AP, TRPC6, CRB2F and AT1), nuclear proteins and transcription factors (WT1, LAMX1B, SMARCL1, NUP93, NUP107, NUP205, XPO5, E2F3, NXF5, PAX2, LAMNA and WDR73), podocyte actin cytoskeletal organization (ACTN4, MYH9, INF2, MYOIE, MAGI2, ANLN, ARHGAP24, ARHGDIA, KANK1/2/4, SYNPO, PTPRO, EMP2, APOL1, CUBN and PODXL), co-enzyme Q biosynthesis (COQ2, COQ6, PDSS2, ADCK4 and MTTL1), lysosomal pathways (SCARB2 and OCRL1), and adhesion to glomerular basement membrane (LAMB2, ITGB4, ITGA3, COL4A 3/4/5), only with one rare exception LAMB2, the protein product of which is enriched in the glomerular basement membrane [18].
Given that the likelihood of detecting a causative gene mutation is inversely related to the age of disease onset, it is advisable to perform clinical genetic testing to those patients showing massive and persistent proteinuria aged under 25 years [19]. Furthermore, the following clinical indications for genetic testing should be taken into consideration: congenital or infantile-onset nephrotic syndrome, childhood-onset nephrotic syndrome with family history, manifesting histologically as FSGS or diffuse mesangial sclerosis, and/or extra-renal manifestations.
Because immune dysregulation has been implicated in the pathogenesis of steroid- SSNS, an exome-wide study within a carefully phenotyped cohort of children identified the human MHC gene HLA-DOQ1 as a risk allele for SSNS [20]. So far, only one causative gene (EMP2) has been discovered, and these findings reinforce the role of adaptive immunity in the etiology of SSNS [21].
More recently, by performing whole exome sequencing (WES) in a cohort of individuals in 17 families with partial glucocorticoid treatment sensitivity, six novel recessive mutations of MAGI2, TNS2, DLCI, CDK20, ITSN1 and ITSN2 have been identified. These six nephrosis genes delineate a pathogenic pathway related to podocytic regulation of Rho-like small GTPase (RLSG) activity, which may be at the intersection between steroid sensitivity and steroid resistance in nephrotic syndrome [22]. At present, corticosteroid immunosuppressive drugs are still adopted as the fist-line treatment without discrimination in patients who are steroid-resistant or steroid-sensitive. Undoubtedly, a definitive molecular diagnosis based on high-throughput massively parallel sequencing could guide us to a more rational treatment decision for these patients. In view of the high proportion of SSNS, timely corticosteroid therapy is necessary. Once the children identified as SRNS, especially for young patients (< 2 years old), genetic testing can be carried out according to the local conditions.
The clinical and genomic spectrum of nephrotic syndrome
In the past, renal histopathology has been used as a crucial criterion for diagnostic, prognostic and therapeutic decisions in children with nephrotic syndrome. However, the selection of treatment strategies based on merely renal histopathological findings is deemed to be flawed in real-world clinical practice [23, 24]. According to the PodoNet Registry cohort report in 2015 [25], 1665 patients were enrolled, including childhood-onset steroid-resistant (age ≤ 20 years old), congenital nephrotic syndrome (CNS), or persistent subnephrotic proteinuria of likely genetic origin. The most common histopathological diagnoses were FSGS (56%), MCD (21%), mesangioproliferative glomerulonephritis (MesPGN, 12%), and diffuse mesangial sclerosis (DMS). Patients with FSGS, MCD, and MesPGN presented with a similar degree of hypoalbuminemia and comparable prevalence of hypotension. Mutations in NPHS2 (n = 138), WT1 (n = 48), and NPHS1 (n = 41) were most commonly identified. Among the various intensified immunosuppressive therapy protocols, calcineurin inhibitors and rituximab yielded consistently high response rates, with 40–45% of patients achieving complete remission. The initial histopathologic diagnosis did not predict the outcome of the intensified immunosuppressive therapies, and the remission rates differed marginally between MCD (51%), MesPGN (40.6%), and FSGS (39.0%). Genetic abnormalities were found in 22% of patients with FSGS, 19% with MesPGN, and 12% with MCD, and the kidney biopsies showed their limited value in distinguishing genetic from non-genetic disease etiologies. Close association with specific genetic disorders was limited to DMS (WT1 and PLCE1) and CNS (NPHS1). It was the genetic diagnosis, but not the histopathologic disease type, that strongly predicated the responsiveness to intensified immunosuppressive therapy.
In the clinical setting, the spectrum of the pathological phenotype in nephrotic syndrome patients with genomic abnormality is variable. Genetically, nephrotic syndrome can be inherited in an autosomal recessive, autosomal dominant or mitochondrial manner, and also can be as isolated renal disease or as part of a multisystem inherited disorder. Mutations in the same gene and even identical mutations can result in utterly different phenotypes, especially in different ethnicities (e.g., WT1) [7]. However, for those sporadic SRNS, 30–32.3% of patients show a defined mutation [26, 27]. So far, there are no treatment guidelines based on SRNS genetic variants. Although clinical studies indicate that some patients with genetic causes could still respond to cyclosporine A, this therapy may be more effective in non-genetic SRNS as compared with genetic variants of SRNS [28]. Therefore, only when a genetic diagnosis is established can the histopathologic findings help with the prognosis of long-term outcome in children with SRNS. This is confirmed by recently integrative analysis from the PodoNet Registry cohort with 1354 patients enrolled [29]. Among 212 patients whose genetic cause was ascertained (NPHS2 and WT1 mutations accounted for two thirds of these), 85% of them would progress to ESRD within 15 years, and this outcome was significantly poorer than that of children with multidrug-resistant disease who carried no genetic abnormality. For example, a multidrug resistant patient with a genetic diagnosis and given GFR would have a nearly threefold higher ESRD risk when diagnosed with FSGS compared with MCD.
With the advent of next-generation sequencing (NGS), the extensive clinical utilization of genetic testing has been facilitated by rapid and relatively inexpensive sequencing technical innovation, such as WES and whole genome sequencing (WGS). Although there is still not a clear guideline pertaining to mutational screening, evidence from the clinical cohort suggests that genetic testing should be guided by the genetic basis of the disease [30, 31]. Similar to WES and WGS, the gene-panel design should take underlying factors into consideration: histological patterns, onset-stage, extra-renal symptoms, family history, renal function at first presentation, and initial treatment response. Despite recognition of an increasing number of genetic causes within SRNS, only mutations in several genes are frequently identified. Novel genetic causes need to be discovered. Furthermore, considering the diverse clinical heterogeneities, even some patients with a negative initial test result may still have an as yet undetected genetic cause, and this will have potential consequences on choosing a therapeutic approach in the future. The molecular ontology of SRNS in the era of NGS will ultimately help not only to uncover the pathogenic pathways, to redefine diagnostic classification and prognostic assessment, but also to provide targets to guide personalized medical management.
Non-immunological effects of immunosuppressants on podocytes
To date, corticosteroids are still the superior treatment for the first attack of nephrotic syndrome, but other immunosuppressive drugs have to be introduced when confronted with FRNS or SRNS [32]. Considering the relative efficiency to prevent relapses, the burden of complications, and the compliance of patients during treatment, immunosuppression therapy requires a careful balance between risks and benefits, and many of these agents have a narrow therapeutic window and require close monitoring [33].
Immunosuppressive therapies are used after confirming steroid resistance, including intravenous steroid pulses, calcineurin inhibitors (CNIs), mycophenolate mofetil (MMF), CNI combined with MMF, oral or intravenous cyclophosphamide (CPH), and rituximab [34]. Due to the etiological heterogeneity, the responses to immunosuppressants are dramatically discrepant among different patients with nephrotic syndrome. For patients without a podocytic gene defect, the response to immunosuppressive therapies with complete remission is about 60% and partial remission is 19% [28, 35]. The PodoNet cohort confirmed that the efficacy of CNI-based therapies was superior to steroid pulses, cyclophosphamide, and MMF, all of which did not show any therapeutic effect in about 85% of patients as first line therapy [34]. What is more, if patients showed resistance to CNIs, these agents (i.e., steroid pulses, cyclophosphamide, and MMF) would be completely non-efficacious as second- or third-line therapies and should be avoided in SRNS [11, 36]. According to this, CNIs are recommended as initial therapy for children with SRNS [37].
A recent study found that B cell depleting therapy with rituximab could induce complete remission in 44% and partial remission in 15% of children with SRNS [34]. Several case series have suggested that rituximab is effective in some SRNS patients who failed to respond to CNIs [25, 38]. However, evidence from an open-label randomized trial of rituximab failed to show any improvement in 31 children with SRNS, compared 16 children who received CNIs, prednisolone, and two infusions of rituximab with 15 children who received CNIs and prednisolone alone [39]. More recently, a parallel-arm, open-label, randomized clinical trial compared the efficacy of rituximab and tacrolimus among children with SDNS and found that rituximab is superior to tacrolimus in maintaining disease remission and minimizing corticosteroid exposure with good tolerance and low nephrotoxicity. This study suggests that rituximab can be used as first-line corticosteroid-sparing therapy in children with SDNS [40]. There is no conclusion as to whether rituximab or CNIs should be preferentially recommended for children with SDNS or SRNS, but it is indisputable that CNIs and rituximab are attracting more attention in the therapy of pediatric nephrotic syndrome.
The non-immunologic anti-proteinuric action of immunosuppressants
Experimental studies suggest that some immunosuppressants show non-immunologic anti-proteinuric action via a direct glomerular effect on podocyte stabilization. For example, cyclosporine appears to stabilize the actin cytoskeleton by blocking the calcineurin-mediated dephosphorylation of synaptopodin [41]; cyclosporine may ameliorate the injury of podocytes by reducing the intracellular influx of calcium required for activation of calcineurin [42], and cyclosporine also can protect the podocytes from injury through inhibiting the signaling pathway of nuclear factor of activated T cell (NATF) [43]. Rituximab may stabilize the action cytoskeleton and prevent podocyte apoptosis by binding directly on sphingomyelin phosphodiesterase acid-like 3b protein (SMPDL3b), independent of its well characterized activity as a monoclonal antibody for CD20 on B lymphocytes [44]. Besides the cellular mechanism of podocyte-stabilization, cyclosporine also has a hemodynamic (somehow nephrotoxic) effect mediating the reduction of glomerular filtration rate (GFR), thereby reducing proteinuria [45]. Besides their well-known anti-inflammatory and immunosuppressive activity primarily mediated by genomic effects, glucocorticoids may protect podocytes from injury through increasing the phosphorylation of nephrin via the serum- and glucocorticoid-regulated kinase, reducing podocyte apoptosis and increasing the number of podocyte progenitors, and preventing podocyte motility and actin disassembly by modulating the production of cyclin guanosine monophosphate [46]. Furthermore, glucocorticoids may have an anti-apoptotic effect by restoring Bcl-2 expression and reducing p53 levels in cultured podocytes treated with puromycin [47, 48]. These effects might explain another side of anti-proteinuric effects of steroids. Levamisole also showed podocyte protective effects by inducing expression of glucocorticoid receptor (GR) and thus enhancing GR signaling [49]. Thus, the advantage of CNIs and rituximab in nephrotic syndrome is due in part to the protective effect on podocytes, which is independent of immunosuppression.
Although immunosuppressive therapies increase the remission of SRNS, therapy of children with podocyte genetic abnormalities remains a serious challenge. There are some patients with genetic forms of nephrotic syndrome who may achieve partial or complete remission with cyclosporine-based immunosuppressive therapy, which have been confirmed by the results of some anecdotal clinical observations and retrospective studies (Table 2). Genetic abnormalities implicated in these studies include WT1 [52], NPHS1 [50], NPHS2 [34], PLCEI [51], and TRPC6 [42], mutations or mutations in the regulators of RHO-like GTPase (ARHGDIA, KANK1, KANK2 and KANK3) [53]. However, for most genetic SRNS, response to cyclosporine-based immunosuppressive therapy was weak and only restricted to exceptional patients. The majority of children cases with podocyte genetic abnormalities are resistant to current immunosuppressive treatments, including corticosteroids, and have a high risk of developing ESRD [54].
The progression of non-immunosuppressive therapy
In current clinical practice, a shift towards genomic medicine is occurring, wherein genome-wide screening will help not only in unraveling the pathogenic pathways of nephrotic syndrome but also in providing potential targets to guide personalized medical management based on specific genes. It is the recognition of the key role of podocyte injury in nephrotic syndrome that has led to identification of several important molecular pathways that are able to regulate podocyte injury and to innovative drugs aiming to regulate these pathways. This knowledge will offer specifically targeted and effective treatments for nephrotic syndrome. Furthermore, combining hemodynamic targeting medicine such as renin–angiotensin–aldosterone system (RAAS) inhibitors, podocyte-specific metabolic targeting therapy and anti-inflammatory drugs might provide extra benefits beyond single-drug treatments.
Specific gene mutations with potential non-immunosuppressive treatments
Podocytes are terminally differentiated cells with limited replicative capacity and are exquisitely vulnerable to cell stress. As the major component of the glomerular filtration barrier, they support other capillary components by counteracting endocapillary pressure, synthesize cytoskeletal proteins and extracellular matrix components, and undertake some immunological roles. To fulfill all of these tasks, podocytes are particularly dependent on energy supply and are rich in mitochondria. Impairment of oxidative phosphorylation in podocytes results in excessive generation of reactive oxygen species (ROS) and functional and structural abnormalities, which subsequently lead to disruption of the glomerular filtration barrier, proteinuria, and ultimately the development of glomerular sclerotic lesions [59]. Therefore, the peculiarity of CoQ10 deficiency among mitochondrial disorders hints that an effective treatment is available. For instance, if a mutation in one of the genes controlling coenzyme Q10 biosynthesis is detected (e.g., COQ2, COQ6, ADCK4 or PDSS2), experimental treatment with coenzyme Q10 may be justified, because there have been reports on a partial response to Q10 supplementation in childhood SRNS cases with mutations in COQ2, COQ6, and ADCK4. As there is no recommend treatment dosage, it appears that 30–50 mg/kg/day may be an appropriate starting dose, and many patients respond to and tolerate oral supplementation with high dose CoQ10 [60]. Yet, once the kidney disease and neurological damage related to CoQ10-deficiency is well established, the clinical conditions cannot be reversed by the addition of CoQ10 [61]. Likewise, patients with CUBN mutations may respond to vitamin B12 addition, while patients with ARHGDA mutation may respond to a mineralocorticoid receptor antagonist, i.e., eplerenone, through modulation of Rac I-mineralocorticoid interaction [53] (Table 2).
Available drugs for non-immunosuppressive treatment
With our expanding knowledge of the molecular mechanisms associated with the pathogenesis of nephrotic syndrome, a variety of alternative drugs with specific target non-immunological options have been developed for patients with nephrotic syndrome [62]. Although most of these agents are only partially effective, or even have marked adverse effects, these alternative treatments will be addressed systematically and prospectively in future (Table 3).
Galactose
Galactose is postulated to have the ability to prevent the focal sclerosis permeability factor (FSPF) from gaining access to the podocyte by interaction with glomerular glycocalyx in a rat model [63]. It has been proposed to be a potential treatment for nephrotic syndrome. Subsequently, galactose-induced remission of nephrotic syndrome was confirmed by a case report in an adult nephrotic syndrome patient with oral galactose, who was resistant to multiple immunosuppressants and plasmapheresis [64]. Moreover, this observation was also confirmed by other two cases of child patients [65]. Yet, according to a prospective pilot clinical trial aiming to evaluate the efficacy of galactose in 2013, galactose decreased FSPS in children with SRNS but failed to alleviate proteinuria levels among five patients with idiopathic SRNS and two patients with post-transplant FSGS recurrence [72]. On the contrary, a more recent clinical report from the novel therapies in resistant FSGS (FONT2) in 2015 affirmed the efficacy of galactose in reducing proteinuria: 21 eligible patients were assigned to one of the three study arms. While none of seven subjects treated with adalimumab (Humira®, TNF antibody) achieved the primary outcome, two subjects in the galactose treatment arm and two in the standard medical therapy arm (treated with lisinopril, losartan, or atorvastatin) had a 50% reduction in proteinuria without a decline in evaluating GFR. The findings of the FONT2 clinical trial group suggested that further studies of novel therapies for rare glomerular disease such FSGS might benefit from enrollment of patients earlier in the course of their disease [73].
ACTH
Different from the mainstream treatments for nephrotic syndrome, ACTH (adrenocorticotropic hormone) is an immunostimulator by itself and has a long history as old as cortisone for the treatment nephrotic syndrome. There has been increasing use of ACTH in the past two decades. ACTH has shown its efficacy in multiple causes of nephropathy, including MCD, FSGS, and mesangial glomerulonephritis, with a responsive rate varying from 29 to 100% [74, 75]. Recently, ACTH has shown its particular benefits in idiopathic membranous nephropathy [76]. Although the exact mechanism for ACTH has not been fully understood, it is thought to act directly on podocytes via binding to the melanocortin 1 receptor [66, 67]. ACTH is currently taken as a potential treatment for nephrotic syndrome, although there are still some conflicting results according to different studies [77]. Some studies have identified that the potential response rate to ACTH in patients with steroid-resistant FSGS may be less than 30%, and its role as primary therapy has yet to be proven [78]. Thus, multicenter randomized controlled trials are urgently needed to evaluate its efficacy in idiopathic nephrotic syndrome.
Vitamin D analogs and stimulation of the calcium-sensing receptor
Similarly, the use of vitamin D analogs (cinacalcet) to stimulate the calcium-sensing receptor has come into the field of alternative therapy for nephrotic syndrome. Via stimulation of calcium-sensing receptors, cinacalcet can enhance podocyte stability and may have the potential to decrease proteinuria in nephrotic syndrome [68]. In contrast to the positive results from experimental studies and a recent meta-analysis in IgA nephropathy [79], vitamin D supplementation did not reduce the relapse rate in a randomized controlled trail in SSNS patients [80]. So far, there are no available data on cinacalcet. Although definitive conclusions about the use of vitamin D analogs and cinacalcet cannot be drawn, future studies are warranted.
Azithromycin
Recently, there was a case report on the validity of sole azithromycin in nephrotic syndrome, which was based on an earlier observation about an additional positive effect of azithromycin on corticosteroid induction therapy for nephrotic syndrome [81]. This report suggested that azithromycin possibly suppressed disease activity in nephrotic syndrome and reduced proteinuria in subsequent relapses. The authors suggested that azithromycin may possibly improve imbalances of the immune system which may be involved in the activity of nephrotic syndrome, and azithromycin may also have some effects on protein selectivity in renal epithelial cells. Given the rarity, severity, and heterogeneity of this disease involved, more studies are needed to reach definite conclusions about azithromycin use for nephrotic syndrome.
Updating inhibition of the RAAS
As a conservative and reliable non-immunosuppressive approach for nephrotic syndrome, inhibiting the RAAS can modulate renal hemodynamics and thus lead to a decrease in proteinuria through mechanisms of reducing glomerular hyperfiltration and intraglomerular pressure, as well as anti-TGFβ-like properties [69]. Well-designed studies have confirmed that mechanical strain on podocytes would lead to the podocyte-specific overexpression of ANG II type 1 receptor (AT1R), resulting in marked foot process effacement and proteinuria without significant changes in systolic blood pressure [70, 71]. Consequently, the utilization of RAAS inhibitors may play a critical role in mitigating podocyte injury and proteinuria in glomerular disease by inhibiting AT1R-mediated effects [82]. RAAS inhibitors, including angiotensin converting enzyme inhibitors (ACEIs) or type1 angiotensin II receptor blockers (ARBs), are now considered as standard treatments for chronic proteinuric kidney disease including nephrotic syndrome. Both of ACEIs and ARBs are effective at reducing proteinuria in a dose-dependent manner. In the current clinical setting, RAAS inhibitors are often used in combination with other immunosuppressants, yet in some cases of hereditary forms of SRNS, CNIs do not offer an extra therapeutic benefit over RAAS blockade alone [29]. Therefore, patients should be spared the side effects of immunosuppressive therapy. The benefits of combined ACEI and ARB therapy have been suggested by some limited-size studies in nephrotic syndrome with genetic abnormality [83]. However, this combinative utilization is usually associated with potential side effects, such as serious hyperkalemia, which often limits its use. From the long-term experience with RAAS inhibitors, these agents would still be favored for early use in younger patients, especially when they are tolerated well and no better choice except immunosuppressants is available. Furthermore, any novel therapy must add benefit on the background of ACEI or ARB therapy, and these agents should therefore be taken as the mainstay treatment for nephrotic syndrome.
More recently, a phase 2, randomized, double-blind, active-control, dose-escalation study (DUET), using sparsentan, a dual endothelin type A (ETA), and angiotensin II type 1 receptor antagonist, has came to a preliminary conclusion. Patients with FSGS could achieve significantly greater reductions in proteinuria after 8 weeks of sparsentan versus irbesartan, and sparsentan was safe and well tolerated [84]. As an ancillary study of the Nephrotic Syndrome Study Network (NEPTUNE) observational study, DUET had been initiated with a view to evaluate the anti-proteinuric efficacy and long-term safety of sparsentan in patients with primary FSGS, and also to assess the impact in genetic forms of nephrotic syndrome [85]. In the near future, sparsentan will be further evaluated in the DUET study open-label treatment period, and in the phase 3 DUPLEX study, to determine whether it produces sustained reduction in proteinuria and stabilizes kidney function compared with AT1 receptor blockade without undue adverse effects. Positive findings from the phase 3 DUPLEX study would represent a major advance in the management of FSGS and would provide important evidence on whether dual ARBs and endothelin blockade might be an effective therapeutic strategy for SRNS.
The unique art—status of Chinese traditional medicine for nephrotic syndrome
Most traditional herb formulations of Chinese medicine and herb natural products have pleiotropic properties. Stephania tetrandra S. Moore (Fang Ji) and Astragalus membranaceus (Huang Qi) are the most important herbs for treating edema and proteinuria, which are prescribed combined or alone in most traditional formulas [86]. Apart from that, extensive research has identified many active ingredients of herbal medicines with the ability to improve existing immunosuppressive therapies for the treatment of nephrotic syndrome. For example, Wuzhi capsule combined with tacrolimus has been shown not only to significantly reduce the dosage of tacrolimus required to maintain an effective blood concentration but also to result in a higher remission rate and shorter time to achieve partial remission [87, 88]. Unfortunately, due to the low methodological quality and the small number of randomized controlled trials (RCTs), there is currently insufficient evidence for determining whether these interventions should be taken for children with nephrotic syndrome [89]. In the future, the therapeutic effect of traditional Chinese medicine in nephrotic syndrome still needs to be carefully evaluated by some well-designed clinical studies.
Conclusion
For more than 50 years, immunosuppressive therapies have been the mainstay for nephrotic syndrome. However, neither their target cells nor the mechanisms of action in nephrotic syndrome are fully elucidated. Furthermore, long-term use of these medications is fraught with adverse effects, treatment resistance, and loss of response among childhood patients with nephrotic syndrome. With the advance of genomic medicine, more and more intrinsic genetic mechanisms will be discovered. In the past decades, the recognition of the key role of podocyte injury in nephrotic syndrome has inspired researchers to identify several important molecular pathways involved in podocyte injury and to develop new drugs regulating these pathways, which continue to offer specific targets and non-immunosuppressive therapies for nephrotic syndrome. Thus, the new non-immunosuppressive strategies will shed light on the treatment of nephrotic syndrome. Therefore, an optimal algorithm based on personalized immunosuppression should be taken into consideration. Alternative non-immunosuppressants aiming to identifying targets directly in the podocyte with minimal toxicity will also be developed and should have priority to be put into force in a timely manner.
References
Pal A, Kaskel F (2016) History of nephrotic syndrome and evolution of its treatment. Front Pediatr 4:56
(1984) Minimal change nephrotic syndrome in children: deaths during the first 5 to 15 years’ observation. Report of the International Study of Kidney Disease in Children. Pediatrics 73:497–501
Mekahli D, Liutkus A, Ranchin B, Yu A, Bessenay L, Girardin E, Van Damme-Lombaerts R, Palcoux JB, Cachat F, Lavocat MP, Bourdat-Michel G, Nobili F, Cochat P (2009) Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr Nephrol 24:1525–1532
(1978) Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int 13:159–165
Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF (2016) Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol 12:768–776
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K (1998) Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1:575–582
Preston R, Stuart HM, Lennon R (2019) Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol 34:195–210
Fakhouri F, Bocquet N, Taupin P, Presne C, Gagnadoux MF, Landais P, Lesavre P, Chauveau D, Knebelmann B, Broyer M, Grunfeld JP, Niaudet P (2003) Steroid-sensitive nephrotic syndrome: from childhood to adulthood. Am J Kidney Dis 41:550–557
(1981) Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. A Report of the International Study of Kidney Disease in Children. Kidney Int 20:765–771
Koskimies O, Vilska J, Rapola J, Hallman N (1982) Long-term outcome of primary nephrotic syndrome. Arch Dis Child 57:544–548
Tarshish P, Tobin JN, Bernstein J, Edelmann CM Jr (1997) Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol 8:769–776
Lombel RM, Gipson DS, Hodson EM, Kidney Disease: Improving Global Outcomes (2013) Treatment of steroid-sensitive nephrotic syndrome: new guidelines from KDIGO. Pediatr Nephrol 28:415–426
Lombel RM, Hodson EM, Gipson DS, Kidney Disease: Improving Global Outcomes (2013) Treatment of steroid-resistant nephrotic syndrome in children: new guidelines from KDIGO. Pediatr Nephrol 28:415–426
Nourbakhsh N, Mak RH (2017) Steroid-resistant nephrotic syndrome: past and current perspectives. Pediatric Health Med Ther 8:29–37
Tullus K, Webb H, Bagga A (2018) Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc Health 2:880–890
Wiggins RC (2007) The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71:1205–1214
Buscher AK, Weber S (2012) Educational paper: the podocytopathies. Eur J Pediatr 171:1151–1160
Hasselbacher K, Wiggins RC, Matejas V, Hinkes BG, Mucha B, Hoskins BE, Ozaltin F, Nurnberg G, Becker C, Hangan D, Pohl M, Kuwertz-Broking E, Griebel M, Schumacher V, Royer-Pokora B, Bakkaloglu A, Nurnberg P, Zenker M, Hildebrandt F (2006) Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 70:1008–1012
Lovric S, Ashraf S, Tan W, Hildebrandt F (2016) Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31:1802–1813
Gbadegesin RA, Winn MP, Smoyer WE (2013) Genetic testing in nephrotic syndrome—challenges and opportunities. Nat Rev Nephrol 9:179–184
Gee HY, Ashraf S, Wan X, Vega-Warner V, Esteve-Rudd J, Lovric S, Fang H, Hurd TW, Sadowski CE, Allen SJ, Otto EA, Korkmaz E, Washburn J, Levy S, Williams DS, Bakkaloglu SA, Zolotnitskaya A, Ozaltin F, Zhou W, Hildebrandt F (2014) Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 94:884–890
Ashraf S, Kudo H, Rao J, Kikuchi A, Widmeier E, Lawson JA, Tan W, Hermle T, Warejko JK, Shril S, Airik M, Jobst-Schwan T, Lovric S, Braun DA, Gee HY, Schapiro D, Majmundar AJ, Sadowski CE, Pabst WL, Daga A, van der Ven AT, Schmidt JM, Low BC, Gupta AB, Tripathi BK, Wong J, Campbell K, Metcalfe K, Schanze D, Niihori T, Kaito H, Nozu K, Tsukaguchi H, Tanaka R, Hamahira K, Kobayashi Y, Takizawa T, Funayama R, Nakayama K, Aoki Y, Kumagai N, Iijima K, Fehrenbach H, Kari JA, El Desoky S, Jalalah S, Bogdanovic R, Stajic N, Zappel H, Rakhmetova A, Wassmer SR, Jungraithmayr T, Strehlau J, Kumar AS, Bagga A, Soliman NA, Mane SM, Kaufman L, Lowy DR, Jairajpuri MA, Lifton RP, Pei Y, Zenker M, Kure S, Hildebrandt F (2018) Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun 9:1960
Sampson MG, Hodgin JB, Kretzler M (2015) Defining nephrotic syndrome from an integrative genomics perspective. Pediatr Nephrol 30:51–63 quiz 59
Anders HJ, Jayne DR, Rovin BH (2016) Hurdles to the introduction of new therapies for immune-mediated kidney diseases. Nat Rev Nephrol 12:205–216
Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, Ksiazek J, Remuzzi G, Erdogan O, Akman S, Dusek J, Davitaia T, Ozkaya O, Papachristou F, Firszt-Adamczyk A, Urasinski T, Testa S, Krmar RT, Hyla-Klekot L, Pasini A, Ozcakar ZB, Sallay P, Cakar N, Galanti M, Terzic J, Aoun B, Caldas Afonso A, Szymanik-Grzelak H, Lipska BS, Schnaidt S, Schaefer F, PodoNet Consortium (2015) Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 10:592–600
Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, Nabhan MM, Kerecuk L, Hegde S, Hughes D, Marks S, Feather S, Jones C, Webb NJ, Ognjanovic M, Christian M, Gilbert RD, Sinha MD, Lord GM, Simpson M, Koziell AB, Welsh GI, Saleem MA (2017) Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 91:937–947
Giglio S, Provenzano A, Mazzinghi B, Becherucci F, Giunti L, Sansavini G, Ravaglia F, Roperto RM, Farsetti S, Benetti E, Rotondi M, Murer L, Lazzeri E, Lasagni L, Materassi M, Romagnani P (2015) Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol 26:230–236
Buscher AK, Beck BB, Melk A, Hoefele J, Kranz B, Bamborschke D, Baig S, Lange-Sperandio B, Jungraithmayr T, Weber LT, Kemper MJ, Tonshoff B, Hoyer PF, Konrad M, Weber S, German Pediatric Nephrology Association (2016) Rapid response to cyclosporin a and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 11:245–253
Trautmann A, Schnaidt S, Lipska-Zietkiewicz BS, Bodria M, Ozaltin F, Emma F, Anarat A, Melk A, Azocar M, Oh J, Saeed B, Gheisari A, Caliskan S, Gellermann J, Higuita LMS, Jankauskiene A, Drozdz D, Mir S, Balat A, Szczepanska M, Paripovic D, Zurowska A, Bogdanovic R, Yilmaz A, Ranchin B, Baskin E, Erdogan O, Remuzzi G, Firszt-Adamczyk A, Kuzma-Mroczkowska E, Litwin M, Murer L, Tkaczyk M, Jardim H, Wasilewska A, Printza N, Fidan K, Simkova E, Borzecka H, Staude H, Hees K, Schaefer F, PodoNet Consortium (2017) Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol 28:3055–3065
Bensimhon AR, Williams AE, Gbadegesin RA (2018) Treatment of steroid-resistant nephrotic syndrome in the genomic era. Pediatr Nephrol. https://doi.org/10.1007/s00467-018-4093-1
Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, Lovric S, Ashraf S, Rao J, Hermle T, Jobst-Schwan T, Widmeier E, Majmundar AJ, Schneider R, Gee HY, Schmidt JM, Vivante A, van der Ven AT, Ityel H, Chen J, Sadowski CE, Kohl S, Pabst WL, Nakayama M, Somers MJG, Rodig NM, Daouk G, Baum M, Stein DR, Ferguson MA, Traum AZ, Soliman NA, Kari JA, El Desoky S, Fathy H, Zenker M, Bakkaloglu SA, Muller D, Noyan A, Ozaltin F, Cadnapaphornchai MA, Hashmi S, Hopcian J, Kopp JB, Benador N, Bockenhauer D, Bogdanovic R, Stajic N, Chernin G, Ettenger R, Fehrenbach H, Kemper M, Munarriz RL, Podracka L, Buscher R, Serdaroglu E, Tasic V, Mane S, Lifton RP, Braun DA, Hildebrandt F (2018) Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 13:53–62
Deschênes G, Dossier C, Hogan J (2018) Treating the idiopathic nephrotic syndrome: are steroids the answer? Pediatr Nephrol. https://doi.org/10.1007/s00467-018-3963-x
Jefferson JA (2018) Complications of immunosuppression in glomerular disease. Clin J Am Soc Nephrol 13:1264–1275
Trautmann A, Lipska-Zietkiewicz BS, Schaefer F (2018) Exploring the clinical and genetic spectrum of steroid resistant nephrotic syndrome: the PodoNet Registry. Front Pediatr 6:200
Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, Kuwertz-Broking E, Wingen AM, John U, Kemper M, Monnens L, Hoyer PF, Weber S, Konrad M (2010) Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 5:2075–2084
de Mello VR, Rodrigues MT, Mastrocinque TH, Martins SP, de Andrade OV, Guidoni EB, Scheffer DK, Martini Filho D, Toporovski J, Benini V (2010) Mycophenolate mofetil in children with steroid/cyclophosphamide-resistant nephrotic syndrome. Pediatr Nephrol 25:453–460
Hodson EM, Willis NS, Craig JC (2010) Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database Syst Rev 11:CD003594
Hamasaki Y, Yoshikawa N, Hattori S, Sasaki S, Iijima K, Nakanishi K, Matsuyama T, Ishikura K, Yata N, Kaneko T, Honda M, Japanese Study Group of Renal Disease (2009) Cyclosporine and steroid therapy in children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 24:2177–2185
Magnasco A, Ravani P, Edefonti A, Murer L, Ghio L, Belingheri M, Benetti E, Murtas C, Messina G, Massella L, Porcellini MG, Montagna M, Regazzi M, Scolari F, Ghiggeri GM (2012) Rituximab in children with resistant idiopathic nephrotic syndrome. J Am Soc Nephrol 23:1117–1124
Basu B, Sander A, Roy B, Preussler S, Barua S, Mahapatra TKS, Schaefer F (2018) Efficacy of rituximab vs tacrolimus in pediatric corticosteroid-dependent nephrotic syndrome: a randomized clinical trial. JAMA Pediatr 172:757–764
Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P (2008) The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14:931–938
Schlondorff J, Del Camino D, Carrasquillo R, Lacey V, Pollak MR (2009) TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol 296:C558–C569
Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F (2010) Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol 21:1657–1666
Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW 3rd (2011) Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 3:85ra46
Fassi A, Sangalli F, Colombi F, Perico N, Remuzzi G, Remuzzi A (1999) Beneficial effects of calcium channel blockade on acute glomerular hemodynamic changes induced by cyclosporine. Am J Kidney Dis 33:267–275
Ponticelli C, Locatelli F (2018) Glucocorticoids in the treatment of glomerular diseases: pitfalls and pearls. Clin J Am Soc Nephrol 13:815–822
Wada T, Pippin JW, Marshall CB, Griffin SV, Shankland SJ (2005) Dexamethasone prevents podocyte apoptosis induced by puromycin aminonucleoside: role of p53 and Bcl-2-related family proteins. J Am Soc Nephrol 16:2615–2625
Wada T, Pippin JW, Nangaku M, Shankland SJ (2008) Dexamethasone’s prosurvival benefits in podocytes require extracellular signal-regulated kinase phosphorylation. Nephron Exp Nephrol 109:e8–e19
Jiang L, Dasgupta I, Hurcombe JA, Colyer HF, Mathieson PW, Welsh GI (2015) Levamisole in steroid-sensitive nephrotic syndrome: usefulness in adult patients and laboratory insights into mechanisms of action via direct action on the kidney podocyte. Clin Sci 128:883–893
Klaassen I, Ozgoren B, Sadowski CE, Moller K, van Husen M, Lehnhardt A, Timmermann K, Freudenberg F, Helmchen U, Oh J, Kemper MJ (2015) Response to cyclosporine in steroid-resistant nephrotic syndrome: discontinuation is possible. Pediatr Nephrol 30:1477–1483
Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Muller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’Toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nurnberg P, Hildebrandt F (2006) Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38:1397–1405
Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, Caridi G, Seeman T, Tory K, Jankauskiene A, Zurowska A, Szczepanska M, Wasilewska A, Harambat J, Trautmann A, Peco-Antic A, Borzecka H, Moczulska A, Saeed B, Bogdanovic R, Kalyoncu M, Simkova E, Erdogan O, Vrljicak K, Teixeira A, Azocar M, Schaefer F, PodoNet Consortium (2014) Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85:1169–1178
Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, Beck BB, Gribouval O, Zhou W, Diaz KA, Natarajan S, Wiggins RC, Lovric S, Chernin G, Schoeb DS, Ovunc B, Frishberg Y, Soliman NA, Fathy HM, Goebel H, Hoefele J, Weber LT, Innis JW, Faul C, Han Z, Washburn J, Antignac C, Levy S, Otto EA, Hildebrandt F (2013) ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123:3243–3253
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Hildebrandt F, SRNS Study Group (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26:1279–1289
Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, Hanevold C, Thies J, Van Hove JLK, Hingorani SR, Lam C (2018) COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol 33:1257–1261
Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, Moon KC, Seong MW, Gwon TR, Park SS, Cheong HI (2017) COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am J Kidney Dis 70:139–144
Rahman S, Clarke CF, Hirano M (2012) 176th ENMC International Workshop: diagnosis and treatment of coenzyme Q(1)(0) deficiency. Neuromuscul Disord 22:76–86
Atmaca M, Gulhan B, Korkmaz E, Inozu M, Soylemezoglu O, Candan C, Bayazit AK, Elmaci AM, Parmaksiz G, Duzova A, Besbas N, Topaloglu R, Ozaltin F (2017) Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr Nephrol 32:1369–1375
Acosta MJ, Vazquez Fonseca L, Desbats MA, Cerqua C, Zordan R, Trevisson E, Salviati L (2016) Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta 1857:1079–1085
Emma F, Montini G, Parikh SM, Salviati L (2016) Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 12:267–280
Montini G, Malaventura C, Salviati L (2008) Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 358:2849–2850
Kemper MJ, Valentin L, van Husen M (2017) Difficult-to-treat idiopathic nephrotic syndrome: established drugs, open questions and future options. Pediatr Nephrol
Savin VJ, McCarthy ET, Sharma R, Charba D, Sharma M (2008) Galactose binds to focal segmental glomerulosclerosis permeability factor and inhibits its activity. Transl Res 151:288–292
De Smet E, Rioux JP, Ammann H, Deziel C, Querin S (2009) FSGS permeability factor-associated nephrotic syndrome: remission after oral galactose therapy. Nephrol Dial Transplant 24:2938–2940
Kopac M, Meglic A, Rus RR (2011) Partial remission of resistant nephrotic syndrome after oral galactose therapy. Ther Apher Dial 15:269–272
Lindskog A, Ebefors K, Johansson ME, Stefansson B, Granqvist A, Arnadottir M, Berg AL, Nystrom J, Haraldsson B (2010) Melanocortin 1 receptor agonists reduce proteinuria. J Am Soc Nephrol 21:1290–1298
Gong R (2014) Leveraging melanocortin pathways to treat glomerular diseases. Adv Chronic Kidney Dis 21:134–151
Oh J, Beckmann J, Bloch J, Hettgen V, Mueller J, Li L, Hoemme M, Gross ML, Penzel R, Mundel P, Schaefer F, Schmitt CP (2011) Stimulation of the calcium-sensing receptor stabilizes the podocyte cytoskeleton, improves cell survival, and reduces toxin-induced glomerulosclerosis. Kidney Int 80:483–492
Perico N, Benigni A, Remuzzi G (2008) Present and future drug treatments for chronic kidney diseases: evolving targets in renoprotection. Nat Rev Drug Discov 7:936–953
Hoffmann S, Podlich D, Hahnel B, Kriz W, Gretz N (2004) Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol 15:1475–1487
Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ (2004) Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 65:30–39
Sgambat K, Banks M, Moudgil A (2013) Effect of galactose on glomerular permeability and proteinuria in steroid-resistant nephrotic syndrome. Pediatr Nephrol 28:2131–2135
Trachtman H, Vento S, Herreshoff E, Radeva M, Gassman J, Stein DT, Savin VJ, Sharma M, Reiser J, Wei C, Somers M, Srivastava T, Gipson DS (2015) Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol 16:111
Berg AL, Arnadottir M (2004) ACTH-induced improvement in the nephrotic syndrome in patients with a variety of diagnoses. Nephrol Dial Transplant 19:1305–1307
Hogan J, Bomback AS, Mehta K, Canetta PA, Rao MK, Appel GB, Radhakrishnan J, Lafayette RA (2013) Treatment of idiopathic FSGS with adrenocorticotropic hormone gel. Clin J Am Soc Nephrol 8:2072–2081
Hladunewich MA, Cattran D, Beck LH, Odutayo A, Sethi S, Ayalon R, Leung N, Reich H, Fervenza FC (2014) A pilot study to determine the dose and effectiveness of adrenocorticotrophic hormone (H.P. Acthar(R) Gel) in nephrotic syndrome due to idiopathic membranous nephropathy. Nephrol Dial Transplant 29:1570–1577
Lieberman KV, Pavlova-Wolf A (2017) Adrenocorticotropic hormone therapy for the treatment of idiopathic nephrotic syndrome in children and young adults: a systematic review of early clinical studies with contemporary relevance. J Nephrol 30:35–44
Kittanamongkolchai W, Cheungpasitporn W, Zand L (2016) Efficacy and safety of adrenocorticotropic hormone treatment in glomerular diseases: a systematic review and meta-analysis. Clin Kidney J 9:387–396
Deng J, Zheng X, Xie H, Chen L (2017) Calcitriol in the treatment of IgA nephropathy with non-nephrotic range proteinuria: a meta-analysis of randomized controlled trials. Clin Nephrol 2017:21–27
Banerjee S, Basu S, Sengupta J (2013) Vitamin D in nephrotic syndrome remission: a case-control study. Pediatr Nephrol 28:1983–1989
Hara H, Hirano D (2018) Azithromycin suppressed relapses of idiopathic nephrotic syndrome in a child. Clin Kidney J 11:54–55
Whaley-Connell A, Nistala R, Habibi J, Hayden MR, Schneider RI, Johnson MS, Tilmon R, Rehmer N, Ferrario CM, Sowers JR (2010) Comparative effect of direct renin inhibition and AT1R blockade on glomerular filtration barrier injury in the transgenic Ren2 rat. Am J Physiol Renal Physiol 298:F655–F661
Hodson EM, Wong SC, Willis NS, Craig JC (2016) Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database Syst Rev 10:CD003594
Trachtman H, Nelson P, Adler S, Campbell KN, Chaudhuri A, Derebail VK, Gambaro G, Gesualdo L, Gipson DS, Hogan J, Lieberman K, Marder B, Meyers KE, Mustafa E, Radhakrishnan J, Srivastava T, Stepanians M, Tesar V, Zhdanova O, Komers R, Group DS (2018) DUET: A Phase 2 Study evaluating the efficacy and safety of sparsentan in patients with FSGS. J Am Soc Nephrol 29:2745–2754
Komers R, Gipson DS, Nelson P, Adler S, Srivastava T, Derebail VK, Meyers KE, Pergola P, MacNally ME, Hunt JL, Shih A, Trachtman H (2017) Efficacy and safety of sparsentan compared with irbesartan in patients with primary focal segmental glomerulosclerosis: randomized, controlled trial design (DUET). Kidney Int Rep 2:654–664.
Wang XQ,Wang L, Tu YC, Zhang YC (2018) Traditional Chinese medicine for refractory nephrotic syndrome: strategies and promising treatments. Evid Based Complement Alternat Med 2018: 8746349
Wei H, Tao X, Di P, Yang Y, Li J, Qian X, Feng J, Chen W (2013) Effects of traditional Chinese medicine Wuzhi capsule on pharmacokinetics of tacrolimus in rats. Drug Metab Dispos 41:1398–1403.
Sun Z, Ren M, Wu Q, Du X (2014) Co-administration of Wuzhi capsules and tacrolimus in patients with idiopathic membranous nephropathy: clinical efficacy and pharmacoeconomics. Int Urol Nephrol 46:1977–1982
Zhong Y, Menon MC, Deng Y, Chen Y, He JC (2015) Recent advances in traditional Chinese medicine for kidney disease. Am J Kidney Dis 66:513–515
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, J., Liu, Z. Treatment of nephrotic syndrome: going beyond immunosuppressive therapy. Pediatr Nephrol 35, 569–579 (2020). https://doi.org/10.1007/s00467-019-04225-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-019-04225-7