
Abstract
Background
Autosomal recessive polycystic kidney disease (ARPKD) is genetically one of the least heterogeneous ciliopathies, resulting primarily from mutations of PKHD1. Nevertheless, 13–20% of patients diagnosed with ARPKD are found not to carry PKHD1 mutations by sequencing. Here, we assess whether PKHD1 copy number variations or second locus mutations explain these cases.
Methods
Thirty-six unrelated patients with the clinical diagnosis of ARPKD were screened for PKHD1 point mutations and copy number variations. Patients without biallelic mutations were re-evaluated and screened for second locus mutations targeted by the phenotype, followed, if negative, by clinical exome sequencing.
Results
Twenty-eight patients (78%) carried PKHD1 point mutations, three of whom on only one allele. Two of the three patients harbored in trans either a duplication of exons 33–35 or a large deletion involving exons 1–55. All eight patients without PKHD1 mutations (22%) harbored mutations in other genes (PKD1 (n = 2), HNF1B (n = 3), NPHP1, TMEM67, PKD1/TSC2). Perinatal respiratory failure, a kidney length > +4SD and early-onset hypertension increase the likelihood of PKHD1-associated ARPKD. A patient compound heterozygous for a second and a last exon truncating PKHD1 mutation (p.Gly4013Alafs*25) presented with a moderate phenotype, indicating that fibrocystin is partially functional in the absence of its C-terminal 62 amino acids.
Conclusions
We found all ARPKD cases without PKHD1 point mutations to be phenocopies, and none to be explained by biallelic PKHD1 copy number variations. Screening for copy number variations is recommended in patients with a heterozygous point mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) belongs to the family of primary cilia-related diseases. Though more than 70 genes have been identified in ciliopathies [1], the genetic homogeneity of ARPKD makes it unique: it is principally caused by mutations of PKHD1 [2]. Recently, mutations of a second gene, DZIP1L, have been identified [3] and promoter mutation of PMM2 was recently described in polycystic kidney disease and hyperinsulinemic hypoglycemia [4]. PKHD1 is expressed in the renal collecting ducts and in the hepatic bile ducts. The encoded protein, fibrocystin, has a receptor-like structure, and plays a role in maintaining calcium homeostasis [5]. Fibrocystin interacts with polycystin-2, a calcium channel, and is also regulated by polycystin-1 [6,7,8,9]. Their role in a common pathway seems to explain the similar renal manifestations of patients with biallelic PKD1 or PKD2 mutations [10, 11]. The expression of fibrocystin—besides other ciliary proteins—is regulated by HNF1B. Its haploinsufficiency is a common cause of cystic kidney dysplasia, diagnosed often as a hyperechogenic kidney in utero, which can also mimic perinatally a mild form of ARPKD [12,13,14]. The clinical presentation of severe ARPKD is highly specific: it is typically diagnosed in utero, with oligohydramnios and extremely enlarged, hyperechogenic kidneys. Thirty percent of the affected children die perinatally because of secondary pulmonary hypoplasia [15]. The diagnosis of mild cases can however be challenging [15]. In these cases, kidneys can be even normal sized, though not smaller than the height-matched median [16]. Hepatic fibrosis (HF) is an obligate feature in ARPKD, secondary to defective remodeling of the ductal plate and hyperplastic biliary ducts [17,18,19]. Nevertheless, its severity is highly variable, and in most cases, it is difficult to detect by ultrasound scan [18,19,20]. The association of hepatic fibrosis to cystic kidneys is common in ciliopathies, most typically in NPHP3- and TMEM67-associated nephronophthisis [21, 22]. Since these can also result in above-average sized hyperechogenic kidneys, their clinical differential diagnosis can also be challenging. Overlapping phenotypes secondary to the influence of second loci or epigenetic factors can further complicate the genetic diagnosis [16, 23, 24].
Genetic confirmation of the diagnosis is demanding for several reasons: PKHD1 is one of the largest genes consisting of 67 exons in its largest transcript, with more than 700 known mutations (URL: http://www.humgen.rwth-aachen.de) [25]. Furthermore, PKHD1 copy number variations (CNVs), which are difficult to detect by sequencing, have been described in some cases [15, 25,26,27]. Finally, the cumulative allele frequency of its mutations is the highest among all ciliopathy genes, resulting in a carrier frequency of 1.5% in the Caucasian population [28]. Therefore, a heterozygous PKHD1 mutation is a relatively common incidental finding, and does not necessarily mean the trans-association of an undetected mutation in an affected patient.
Despite the principal role of PKHD1 in ARPKD, no PKHD1 mutations are identified in 13–20% of the cases by sequencing [15, 16, 27, 29,30,31,32,33]. Here we wanted to find the reason for these cases: whether they result from undetected PKHD1 mutations or from second locus mutations. We therefore aimed to identify the causal mutations in all cases within a cohort of 36 patients diagnosed with ARPKD by sequencing and multiplex ligation-dependent probe amplification (MLPA) analysis of PKHD1, followed, if negative, by mutational screening of second locus mutations and by clinical exome sequencing. We show that the cases negative for PKHD1 point mutations are phenocopies.
Materials and methods
Patients
Thirty-six unrelated patients from four Hungarian pediatric nephrology centers were included, based on the following criteria: (1) hyperreflective kidneys with microcysts (< 2 cm in diameter) on ultrasound, (2) a kidney length above the 50th percentile (http://radiology-universe.org/calculator/pediatric-kidney-sizes/calculator.php) on at least one side, (3) a transmission compatible with autosomal recessive inheritance, (4) no urinary tract malformation and (5) no extra-renal and -hepatic involvement suggestive of other ciliopathies. None of the families was known to be consanguineous. Parents and patients gave informed written consent.
Screening for PKHD1 mutations
Genomic DNA was isolated from peripheral blood by standard methods. The exons and intronic junctions were amplified using the primers described by Losekoot et al. [34] and Sanger sequenced on an ABI Prism 310 Genetic Analyzer (Thermo Fischer Scientific, Waltham, MA, USA). Patients without biallelic point mutations were subsequently screened for CNVs by MLPA, performed according to the manufacturer’s instructions (MRC-Holland, Amsterdam, the Netherlands). Briefly, DNA was denatured and hybridized overnight with the probe mixes P341 and P342. Hybridized probes were ligated, and amplified with 5′-labeled fluorescent primers. Following separation on an ABI Prism 310 Genetic Analyzer, copy numbers were calculated based on the normalized peak heights following intra- and intersample normalization. Parental samples were screened for the identified mutations to confirm segregation and trans-heterozygosity.
Screening for second locus CNVs
Patients without biallelic PKHD1 mutations were re-evaluated based on their most recent phenotype, and were screened for second locus mutations accordingly. Deletions of NPHP1 and HNF1B were tested by QMPSF analysis as described previously [12, 35]. Similarly, continuous gene deletion of TSC2 and PKD1 was screened by QMPSF analysis, with the primers listed in Suppl. Table 1, according to the protocol of NPHP1-QMPSF [35] with the differences detailed in Suppl. Methods. The deletion of PKD1 was validated with MLPA, using the SALSA MLPA probemix P352 PKD1-PKD2, performed according to the manufacturer’s instructions (MRC-Holland, Amsterdam, the Netherlands).
Screening for second locus (PKD1, PKD2, HNF1B and TMEM67) small-scale mutations
Coding exons and intronic junctions of HNF1B and TMEM67 were Sanger sequenced. A dominant polycystic kidney panel was designed using the Ion AmpliSeq Designer version 4.2.1 (Thermo Fischer Scientific, Waltham, MA). Sample enrichment was performed by the Ion AmpliSeq Library Kit (Thermo Fischer Scientific). Samples were barcoded and sequenced on IonTorrent 316 chip (Thermo Fischer Scientific). More than 95% of the target sequence was covered at least 50-fold. Putative disease-causing genetic variants were validated by Sanger sequencing.
Clinical exome sequencing
Clinical exome sequencing was performed as described previously [36]. More than 95% of the target sequence was covered at least 20-fold. Reads were mapped against the human NCBI37/hg19 reference genome. DNA alignment and sequence variant analysis were carried out using NextGene Software version 2.4.2 (SoftGenetics, State College, PA). Data from the proband was filtered for coding and splicing region mutations of known/putative genes associated with cystic kidney diseases.
Results
PKHD1 mutations
Of the 36 unrelated patients with a clinical diagnosis of ARPKD, 27 (75%) were found to carry biallelic PKHD1 mutations (Table 1). Among them, 25 patients carried biallelic point mutations (P1–25) and two were compound heterozygous for a point mutation and either a duplication of exons 33–35 (P26) or a large deletion encompassing exons 1 to 55 (P27). Furthermore, one patient (P28) was found to carry a single heterozygous frameshift mutation. No PKHD1 mutation was found in 8 families (Table 2, Fig. 1).

Mutational screening procedure in children with the clinical diagnosis of autosomal recessive polycystic kidney disease (ARPKD)
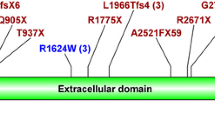
Two mutations, p.Thr36Met and p.Ser2639* were found frequently, in 15/54 (28%) and in 8/54 (15%) of the mutated alleles, respectively. Seven mutations were novel: besides the duplication of exons 33–35 and the deletion of exons 1–55, three truncating mutations (c.5_8delCTGC, p.Ala3Glyfs*2; c.5088delTG, p.Gly1696fs*1; c.12036delA, p.Gly4013Alafs*24) and two missense mutations (c.4328G > A, p.Cys1443Tyr and c.10621A > T, p.Asn3541Tyr). We considered these two latter to also be pathogenic, because neither is reported in the gnomAD database, both affect amino acids conserved in mammals and are predicted to be pathogenic by Polyphen-2 (score: 1.0 and 0.981, respectively) and MutationTaster.
Second locus mutations
All eight families without PKHD1 mutations were found to carry second locus mutations (Table 2, Fig. 1). Three children diagnosed with hyperechogenic, normal-sized kidneys in utero or in infancy carried a de novo deletion of HNF1B. The renal morphology of two children became suggestive of ADPKD between 2 and 4 years of age. They both harbored de novo PKD1 mutations. One patient (P31) was diagnosed with tuberous sclerosis at the age of 4 years, and carried a de novo TSC2/PKD1 deletion. Finally, the phenotype of a patient (P35) diagnosed at the age of 11 years with end-stage renal disease was suggestive of juvenile nephronophthisis. He was compound heterozygous for a complete and a partial deletion of NPHP1, as described recently [35]. A sibling pair (P36, P37), diagnosed with hyperechogenic kidneys and hepatic fibrosis, was homozygous for the frequent TMEM67 missense mutation, p.Cys615Arg [21] (for the calculations, genotype information from only one of them was used). They had no neurological involvement. The patient with a heterozygous PKHD1 mutation (P28) was not found to carry a second locus mutation, even by clinical exome sequencing, which also well covered the coding regions of NPHP2, NPHP3, and WDR19/NPHP13. The mutational screening procedure is summarized in Fig. 1.
Phenotype of patients with PKHD1 and second locus mutations
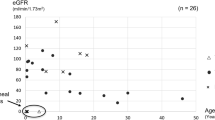
Of the 27 patients with PKHD1 mutations, 19 children (70%) developed perinatal respiratory failure, and nine (33%) died perinatally (< 3 months of age) (Table 1). In contrast, only one of the nine patients (from eight families) with second locus mutations had a transient perinatal respiratory failure, secondary to an infection (Table 2). Among the perinatal cases who survived beyond that period, all but one of the ten children with PKHD1 mutations developed hypertension by the age of 1 year, compared with none of the six patients with second locus mutations (Table 1). A mean kidney length above + 4 SD at diagnosis was also specific for PKHD1-associated ARPKD: 19 of 27 patients (70%) with biallelic PKHD1 mutations, but none of the nine patients with second locus mutations had such an enlarged kidney. Based on these data, the phenotype of the patient with a single heterozygous PKHD1 mutation (P28) is highly suggestive of a moderate PKHD1-associated ARPKD: she developed hypertension at an early stage, as well as highly enlarged kidneys and hepatic fibrosis. There was no difference in the renal survival between patients with PKHD1 mutations who survived the perinatal period and the heterogeneous group of patients with second locus mutations (Tables 1 and 2, Fig. 2).

Renal survival of patients with PKHD1 and second locus mutations. No difference was found in the renal progression between patients with PKHD1 and second locus mutations (p = 0.51). Among patients with second locus mutations, those with PKD1 and HNF1B mutations were young (≤ 11 years), and patients with nephronophthisis reached end-stage renal disease between 9 and 19 years of age, giving a worse renal survival curve than generally expectable
Discussion
Here we aimed to investigate the reason for the high proportion of ARPKD cases negative for PKHD1 point mutations. Our PKHD1-positive rate of 78%—including a patient with a single heterozygous mutation—corresponds well to previous reports [15, 16, 27, 29, 32, 33]. We first assessed the potential role of PKHD1 CNVs, but found no biallelic CNVs in the PKHD1 negative cases. This finding was in accordance with the low prevalence of PKHD1 CNVs in other cohorts [15, 16, 26, 27, 30, 31]. We only found a three-exon duplication and a large deletion in two out of three patients with a single heterozygous PKHD1 point mutation. Duplications are extremely rare; here, we present only the second such case [26]. These results emphasize that screening for CNVs is primarily important in patients with a heterozygous PKHD1 point mutation.
To identify all mutations of the coding regions, we used the most sensitive but also labor- and time-consuming methods of Sanger sequencing and MLPA analysis. However, excluding the causal role of PKHD1 intronic and promoter mutations or rearrangements in patients with or without a single heterozygous mutation is especially challenging by direct genetic tests. Therefore, we aimed to identify the causal mutations in all patients with a clinical diagnosis of ARPKD, and thus exclude the causal role of PKHD1 indirectly. We found all patients without PKHD1 mutations to carry causal mutations in second loci, by re-evaluation of the phenotype and targeted mutation screening, indicating that 22% of the initial clinical ARPKD diagnoses were false. We failed to identify in the third child with a heterozygous PKHD1 mutation a second locus mutation, even following clinical exome sequencing that also covered the promoter region of PMM2 [4]. This is in accordance with the phenotype which is strongly suggestive of PKHD1-associated ARPKD. Her case thus points on the difficulty in identifying some PKHD1 mutations even by the combined approach of sequencing and MLPA, and suggests a potential role of an intronic or a regulatory PKHD1 mutation.
The successful identification of second locus mutations in negative cases by direct genetic tests emphasizes the importance of their thorough re-phenotyping. The differential diagnosis of mild and moderate forms without extremely enlarged kidneys and respiratory failure can be challenging at diagnosis. Within this cohort, an infant-onset hypertension was highly suggestive of PKHD1-associated ARPKD.
In accordance with the literature, the phenotype of patients with PKHD1 mutations strongly correlated with the causal mutations; no patient survived the perinatal period with biallelic loss-of-function mutations [16, 37, 38]. Interestingly, a patient, compound heterozygous for a second and a last exon truncating mutation, p.Gly4013Alafs*24 (P14), presented with a moderate phenotype: she was diagnosed at the age of 5 months and had a normal GFR at the age of 9 years, indicating that the loss of the C-terminal 62 amino acids of fibrocystin does not cause complete loss of function. The intracellular C-terminal part of fibrocystin consists of 192 amino acids and is known to modulate the mTOR pathway [39]. It also contains the ciliary targeting sequence (p.3876_3893CLVCCWLKRSKSRKTKPE), that remains unaffected in the Gly4013Alafs*24 fibrocystin [40]. Along the same lines as the hypomorphic nature of this C-terminal truncation, mice lacking the last exon (exon 67), which encodes the nuclear localization signal and the polycystin 2 binding domain, develop a normal phenotype [41].
In conclusion, we found one quarter of the ARPKD cases to be phenocopies, caused by second locus mutations. Our data suggest that perinatal respiratory failure, a kidney length > + 4 SD and early-onset hypertension increase the likelihood of PKHD1-associated ARPKD. The phenotype of cases that are negative on PKHD1 sequencing should first be re-evaluated. We recommend screening for PKHD1 CNVs in patients with a heterozygous point mutation and in families with an unequivocal phenotype.
References
Kurschat CE, Muller RU, Franke M, Maintz D, Schermer B, Benzing T (2014) An approach to cystic kidney diseases: the clinician’s view. Nat Rev Nephrol 10:687–699
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30:259–269
Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, Ortiz-Bruchle N, Hilger N, Metzis V, Hiersche M, Tay SY, Tunningley R, Vij S, Courtney AD, Whittle B, Wuhl E, Vester U, Hartleben B, Neuber S, Frank V, Little MH, Epting D, Papathanasiou P, Perkins AC, Wright GD, Hunziker W, Gee HY, Otto EA, Zerres K, Hildebrandt F, Roy S, Wicking C, Bergmann C (2017) Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet 49:1025–1034
Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, Anton-Gamero M, Argente J, Bussell AM, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Perez-Cerda C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering D, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S, Bockenhauer D (2017) Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol 28:2529–2539
Nagano J, Kitamura K, Hujer KM, Ward CJ, Bram RJ, Hopfer U, Tomita K, Huang C, Miller RT (2005) Fibrocystin interacts with CAML, a protein involved in Ca2+ signaling. Biochem Biophys Res Commun 338:880–889
Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP (2006) Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17:178–187
Wu M, Yu S (2016) New insights into the molecular mechanisms targeting tubular channels/transporters in PKD development. Kidney Dis (Basel) 2:128–135
Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM (2005) Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Ren Physiol 289:F978–F988
Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD (2006) Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Ren Physiol 290:F1320–F1328
Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, Rudenhed B, Nasr SH, Torres VE, Torra R, Bogdanova N, Harris PC (2010) Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 21:1097–1102
Losekoot M, Ruivenkamp CA, Tholens AP, Grimbergen JE, Vijfhuizen L, Vermeer S, Dijkman HB, Cornelissen EA, Bongers EM, Peters DJ (2012) Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J Med Genet 49:37–40
Bellanne-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, Dubois-Laforgue D, Dusselier L, Gautier JF, Jadoul M, Laloi-Michelin M, Jacquesson L, Larger E, Louis J, Nicolino M, Subra JF, Wilhem JM, Young J, Velho G, Timsit J (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54:3126–3132
Hiesberger T, Shao X, Gourley E, Reimann A, Pontoglio M, Igarashi P (2005) Role of the hepatocyte nuclear factor-1beta (HNF-1beta) C-terminal domain in Pkhd1 (ARPKD) gene transcription and renal cystogenesis. J Biol Chem 280:10578–10586
Williams SS, Cobo-Stark P, Hajarnis S, Aboudehen K, Shao X, Richardson JA, Patel V, Igarashi P (2014) Tissue-specific regulation of the mouse Pkhd1 (ARPKD) gene promoter. Am J Physiol Ren Physiol 307:F356–F368
Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, Elion J, Grandchamp B, Michel-Calemard L, Missy P, Zaccaria I, Le Nagard H, Gerard B, Loirat C, Societe Francaise de F, Barbet J, Beaufrere AM, Berchel C, Bessieres B, Boudjemaa S, Buenerd A, Carles D, Clemenson A, Dechelotte P, Devisme L, Dijoud F, Esperandieu O, Fallet C, Gonzales M, Hillion Y, Jacob B, Joubert M, Kermanach P, Lallemand A, Laquerriere A, Laurent N, Liprandi A, Loeuillet L, Loget P, Martinovic J, Menez F, Narcy F, Roux JJ, Rouleau-Dubois C, Sinico M, Tantau J, Wann AR (2010) Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int 77:350–358
Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schoneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Buttner R, Zerres K, Apn (2005) Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int 67:829–848
Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T (2013) Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 144:112–121 e112
Shneider BL, Magid MS (2005) Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transplant 9:634–639
Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M (2009) Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol 39:100–111
Zerres K, Mucher G, Becker J, Steinkamm C, Rudnik-Schoneborn S, Heikkila P, Rapola J, Salonen R, Germino GG, Onuchic L, Somlo S, Avner ED, Harman LA, Stockwin JM, Guay-Woodford LM (1998) Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 76:137–144
Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MT, Utsch B, Becker C, Nurnberg G, Nurnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F (2009) Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 46:663–670
Tory K, Rousset-Rouviere C, Gubler MC, Moriniere V, Pawtowski A, Becker C, Guyot C, Gie S, Frishberg Y, Nivet H, Deschenes G, Cochat P, Gagnadoux MF, Saunier S, Antignac C, Salomon R (2009) Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int 75:839–847
Arbeiter A, Buscher R, Bonzel KE, Wingen AM, Vester U, Wohlschlager J, Zerres K, Nurnberger J, Bergmann C, Hoyer PF (2008) Nephrectomy in an autosomal recessive polycystic kidney disease (ARPKD) patient with rapid kidney enlargement and increased expression of EGFR. Nephrol Dial Transplant 23:3026–3029
Rossetti S, Harris PC (2007) Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol 18:1374–1380
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN (2017) The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 136:665–677
Miyazaki J, Ito M, Nishizawa H, Kato T, Minami Y, Inagaki H, Ohye T, Miyata M, Boda H, Kiriyama Y, Kuroda M, Sekiya T, Kurahashi H, Fujii T (2015) Intragenic duplication in the PKHD1 gene in autosomal recessive polycystic kidney disease. BMC Med Genet 16:98
Bergmann C, Kupper F, Schmitt CP, Vester U, Neuhaus TJ, Senderek J, Zerres K (2005) Multi-exon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD). J Med Genet 42:e63
Sweeney WE Jr, Avner ED (2011) Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol 26:675–692
Krall P, Pineda C, Ruiz P, Ejarque L, Vendrell T, Camacho JA, Mendizabal S, Oliver A, Ballarin J, Torra R, Ars E (2014) Cost-effective PKHD1 genetic testing for autosomal recessive polycystic kidney disease. Pediatr Nephrol 29:223–234
Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik-Schoneborn S, Furu L, Onuchic LF, De Baca M, Germino GG, Guay-Woodford L, Somlo S, Moser M, Buttner R, Zerres K (2003) Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol 14:76–89
Obeidova L, Seeman T, Elisakova V, Reiterova J, Puchmajerova A, Stekrova J (2015) Molecular genetic analysis of PKHD1 by next-generation sequencing in Czech families with autosomal recessive polycystic kidney disease. BMC Med Genet 16:116
Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, Somlo S, Germino GG, Guay-Woodford LM (2005) Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet 42:336–349
Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica-Worms K, Bryant J, Bernardini I, Fischer R, Huizing M, Guay-Woodford L, Gahl WA (2010) PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab 99:160–173
Losekoot M, Haarloo C, Ruivenkamp C, White SJ, Breuning MH, Peters DJ (2005) Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum Genet 118:185–206
Javorszky E, Moriniere V, Kerti A, Balogh E, Piko H, Saunier S, Karcagi V, Antignac C, Tory K (2017) QMPSF is sensitive and specific in the detection of NPHP1 heterozygous deletions. Clin Chem Lab Med 55:809–816
Orosz O, Rajta I, Vajas A, Takacs L, Csutak A, Fodor M, Kolozsvari B, Resch M, Senyi K, Lesch B, Szabo V, Berta A, Balogh I, Losonczy G (2017) Myopia and late-onset progressive cone dystrophy associate to LVAVA/MVAVA exon 3 interchange haplotypes of opsin genes on chromosome X. Invest Ophthalmol Vis Sci 58:1834–1842
Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, Germino GG, Guay-Woodford LM, Somlo S (2003) Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol 14:2004–2014
Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, Navarro M, El-Youssef M, Torres VE, Harris PC (2003) A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int 64:391–403
Wang S, Wu M, Yao G, Zhang J, Zhou J (2014) The cytoplasmic tail of FPC antagonizes the full-length protein in the regulation of mTOR pathway. PLoS One 9:e95630
Follit JA, Li L, Vucica Y, Pazour GJ (2010) The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J Cell Biol 188:21–28
Outeda P, Menezes L, Hartung EA, Bridges S, Zhou F, Zhu X, Xu H, Huang Q, Yao Q, Qian F, Germino GG, Watnick T (2017) A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int 92:1130–1144
Funding
This work was supported by OTKA K109076 and Ministry of National Economy, Hungary GINOP-2.3.2-15-2016-00039 (to István Balogh, Zoltán Maróti and Tibor Kalmár), MTA-SE Lendulet Research Grant (LP2015-11/2015) of the Hungarian Academy of Sciences and NKFIA/OTKA K109718, KH125566 (to Kálmán Tory).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Parents and patients gave informed written consent.
Rights and permissions
About this article

Cite this article
Szabó, T., Orosz, P., Balogh, E. et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr Nephrol 33, 1713–1721 (2018). https://doi.org/10.1007/s00467-018-3992-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-018-3992-5