
Abstract
Background
Complement factor B gene (CFB) is an important component of the alternate pathway of complement activation that provides an active subunit that associates with C3b to form the C3 convertase, which is an essential element in complement activation. Among the complement-associated disorders, mutations and pathogenic variants in the CFB gene are relatively rare phenomena. Moreover, mutated CFB affiliation with immune-complex diffuse membranoproliferative glomerulonephritis (IC-MPGN) and atypical hemolytic uremic syndrome (aHUS) are considered a highly rare occurrence.
Case presentation
We describe the clinical presentation, course, and pathological findings in a 7-year-old boy who has confirmed CFB heterozygous variants with pathological features compatible with IC-MPGN. Mutational analysis revealed a heterozygous variant p.Glu566Arg in exon 13 of the CFB gene. The patient did not respond to steroids and mycophenolate mofetil (MMF) therapy but responded clinically and biochemically to eculizumab treatment. This is the first case report of CFB alteration associated with IC-MPGN and aHUS that was successfully treated with eculizumab.
Conclusions
Heterozygous variants in the CFB gene can be pathogenic and associated with IC-MPGN and aHUS. Early diagnosis and prompt management can be essential in preventing end-stage renal disease. Eculizumab may provide an effective modality of treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Complement factor B (CFB) gene is an important component of the alternate pathway of complement activation. Factor B circulates in the blood as a single-chain polypeptide and provides the catalytic subunit of the C3/C5 convertase of the alternate complement pathway [1, 2]. Mutations in complement proteins primarily occur in regulatory proteins like complement factor H (CFH), complement factor H-related proteins (CFHRs), and complement factor I (CFI). In contrast, mutations in the CFB gene are considered rare, and their coalitions with immune complex diffuse membranoproliferative glomerulonephritis (IC-MPGN) are exceedingly rare. It is recognized that activation of the alternate pathway secondary to mutated complement genes could result in dense-deposit disease (DDD) and C3 glomerulonephritis (C3GN), but not in IC-MPGN [3–5]. Moreover, heterozygote variants of the CFB gene are regarded as polymorphisms or have uncertain clinical significance. Herein we describe a patient with a pathogenic variant in the CFB gene who was diagnosed with IC-MPGN and aHUS, which were successfully treated with eculizumab. We also highlight the pathogenicity of CFB heterozygote variant and emphasize the importance of early diagnosis and proper management.
Case presentation
A 7-year-old boy presented on 5 October 2015 with 4-days’ history of fever, worsening edema, dark urine, and decreased urine output. He received a course of antibiotic (amoxicillin) in a private clinic, but his symptoms worsened; therefore, he was referred to our service. His parents were not relatives, and there was no history of renal diseases in parents or siblings. On physical examination, the patient showed generalized edema and elevated blood pressure (142/94 mmHg); other vital signs were normal. Initial laboratory tests revealed renal impairment. His blood urea nitrogen (BUN) was 24.3 mmol/L (normal 2.5–8 mmol/L) and serum creatinine (sCr) 275 μmol/L (44–88 μmol/L) with estimated glomerular filtration (eGFR) rate of 16 ml/min/1.73 m2 (new Schwartz equation). Electrolytes and blood gas analysis were within normal limits, albumin was 22 g/L [38–54 g/L], and complete blood count (CBC) showed hemoglobin of 10.7 g/dl (10–12 g/dl) and normal leukocyte and platelet count of 247 × 109/l (150–400 × 109/l). Lactate dehydrogenase (LDH) was elevated (500 U/L) [125-200 U/L]. Urine analysis showed macroscopic hematuria and nephrotic-range proteinuria (protein creatinine ratio 1600 mg/mmol, normal 30 mg/mmol). Complement 3 (C3) level was low (0.18 g/l; normal [0.8-1.2 g/l), while C4 level was normal. Coombs test was negative, and peripheral blood smear showed no schistocytes. Serology for antinuclear, anti-double-stranded DNA, anti-streptolysin O (ASO), antiphospholipid, anti-glomerular basement membrane (GBM), and antineutrophilic cytoplasmic antibodies were negative. There was no serological evidence of hepatitis B and C or human immunodeficiency viral infections. Throat, blood, and urine cultures were negative. Renal Doppler ultrasound was normal.
The patient underwent renal biopsy. The sample was processed routinely for conventional light microscopy, immunofluorescence (IF), and ultrastructural examinations. Histologically, the biopsy showed diffuse proliferative immune-type crescentic glomerulonephritis (Fig. 1a–c). There were 13 glomeruli, none of which were globally sclerosed. The mesangium was expanded by increased mesangial matrix and diffuse moderate increase in mesangial cellularity. Lobulation of the glomerular tufts and mesangiolysis were evident. GBMs demonstrated an increase in thickness and segmental double contouring on periodic acid-Schiff (PAS) and methenamine silver special stains. Proliferative features in the form of neutrophilic inflammatory infiltrate into the glomerular capillaries, segmental subendothelial deposits, and exuberant cellular crescent formation were present; the latter was identified in ten glomeruli. Marked interstitial edema, moderate interstitial acute and chronic interstitial inflammation, and moderate acute tubular epithelial cell degenerative and regenerative changes were noted. No granulomas or significant eosinophilic infiltrate were identified, and no glomerular or interstitial microthrombi were seen. There was no interstitial fibrosis or tubular atrophy. The renal vasculature was unremarkable, and there was no evidence of vasculitis.

Light, immunofluorescence (IF), and electron microscopy (EM) pathological findings: a–c Light microscopy showing membranoproliferative pattern of glomerular injury. a Marked endocapillary proliferation with large numbers of infiltrating neutrophils and segmental double contouring of the glomerular basement membranes (GBM) (arrows) [periodic acit-Schiff (PAS x 400)]. b Lobulation of glomerular tufts and segmental subendothelial deposits (arrow) (PAS x 400). c Exuberant cellular crescents (PAS x 200): d, e IF shows intense glomerular mesangial and capillary staining for immunoglobulin G (IgG) (d x 400) and C3 (e x 400). c EM micrograph demonstrates subendothelial immune-type dense deposits (transmission electron microscopy, uranyl acetate, and lead citrate stain x 8000)
Immunofluorescence study demonstrated diffuse and global glomerular capillary and mesangial coarse granular staining for immunoglobulin G (IgG) (2+) (Fig. 1d); Kappa (2+) and Lambda (2+) light chains; C3 (3+) (Fig. 1e), and C1q (3+) (not shown) in nonsclerotic glomerular tufts. There was no staining for IgM, IgA, and C4. Ultrastructurally (Fig. 1f), the increased mesangial matrix and hypercellularity, and endocapillary hypercellularity were noted, along with extensive immune-type dense deposits, predominantly at the subendothelial zone. Mesangial and scattered small subepithelial immune-type dense deposits were also present. Foot processes of visceral glomerular epithelial cells showed diffuse effacement. Immunohistochemical staining for C4d (polyclonal, dilution 1:40, Abcam, Cambridge, UK) was negative in glomeruli.
Accordingly, the patient was started on pulse methylprednisolone (10 mg/kg, three doses), followed by prednisolone orally (2 mg/kg per day) and mycophenolate mofetil (MMF) (600 mg/m2/day). Amlodipine was added to control blood pressure. Renal function continued to worsen, and the patient remained oliguric, which necessitated intermittent hemodialysis for 3 weeks. Hemodialysis began on day 25 of admission and continued until day 42of admission. However, during this treatment, he was given adequate challenge of dialysis-free period, but he continued to have poor renal clearance and remained oliguric. On physical examination, the patient showed persistent generalized edema and sustained high blood pressure (138/92 mmHg). Notably, there were no signs of diarrhea or gastrointestinal infection; therefore, testing for Shiga toxin was not done. On day 33 of admission, the patient looked slightly pale and showed signs of hemolysis in the form of thrombocytopenia (108 × 109/l), increased serum LDH (702 U/l) (125–200 U/l) and reduced serum haptoglobin (0.26 g/l) [2.7 g/l], which were compatible with aHUS. His serum C3 continued to be low. Circulating C3 nephritic factor was not detectable. Laboratory parameters are summarized in Table 1.
The persistence of low C3 levels, deteriorating renal function, and evolution of signs of hemolysis warranted starting anti-C5 monoclonal blocking antibody (eculizumab) therapy on day 36 of admission (600 mg weekly for 3 weeks, followed by 600 mg every 2 weeks), as per standard protocol [4]. Following the third dose, he showed significant improvement in renal function, serum C3 level, and urine output (Fig. 2). His immunosuppressant—namely, MMF—was discontinued on the day eculizumab was started; steroids were tapered slowly and stopped on the day 75 of admission.

The above chart clearly represents improving trend of Serum creatinine and serum C3 complement levels
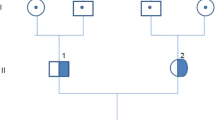
Later, molecular testing revealed a heterozygote variant in exon 13 of CFB, which changes a glutamine at amino acid position 566 to an arginine (c.1697A > C p.Glu566Arg). Furthermore, no mutations were identified in ADAMTS 13, C3, MCP, CFH, CFI, CFHR1-5, DGKE, and THBD genes. Anticomplement factor B antibodies and functional analysis of alternate complement activity were not available in our center. Genetic analysis of family members demonstrated the same heterozygous CFB gene variant in the 52-year-old father, who showed no evidence of renal function impairment, proteinuria, hematuria, or hemolysis. Analysis of the mother and the elder brother and sister was normal.
At 3-months’ follow-up, the patient was well, with eGFR of 100 ml/min/1.73 m2, BUN 6.3 mmol/L, sCr 45 μmol/L, and albumin 39 g/L. Complete blood count showed a hemoglobin of 11.7 g/dl, normal leukocyte count, and platelet count of 267 × 109/l. His proteinuria improved to 96 mg/mmol. As per evidence, discontinuation of eculizumab remains unknown in complement factor B mutation, so our clinical decision is to continue eculizumab every 2 weeks until further confirmatory evidence.
Discussion
Recently, MPGN was reclassified to immune-complex-mediated (IC-MPGN) and complement alternative pathway-mediated (C3 glomerulopathy), which comprises DDD and C3GN. The pattern of glomerular injury and morphological features are similar in these entities, but they have different IF and ultrastructural characteristics that define them pathologically. IC-MPGN can be associated with chronic infections, autoimmune diseases, and presence of circulating immune complexes triggered by activation of classic and terminal complement pathways and characterized by C3 and immunoglobulins deposition on IF, predominantly IgG. However, C3 glomerulopathy is caused by mutations in the complement-regulating proteins, which results in dysregulation of the alternative complement pathway and is characterized by intense IF deposition of C3 (staining intensity of ≥ 2 orders of magnitude greater than any other immune reactant, using a scale from 0 = trace, 1+ to 3+ and without significant staining for immunoglobulins or C1q ) [6, 7].
Most mutations and variants in complement regulatory proteins are associated with C3, CFH, CFI, CFHR1-5 and MCP genes [2, 8, 9]. C3 glomerulonephritis and DDD are commonly associated with mutations in these genes but rarely with alterations in CFB [3–5]. Two cases of symptomatic C3GN associated with CFB mutation were previously reported [10, 11]. Imamura et al. [10] described a 12-year-old girl with a biopsy-proved C3GN associated with heterozygosity c.1099A.C, p.S367R in the CFB gene, p.R201S in the CFI gene, and p.V916I in C3 gene. DNA analysis demonstrated the same alteration in the CFB gene in the patient’s asymptomatic mother and elder brother. Sethi et al [11] reported a 22-year-old woman with C3GN that associated with the heterozygous variant c.1697A.G, p.Glu566Ala (an adenine to guanine change at nucleotide 1697, leading to a substitution of glutamate by alanine at amino acid 566). In both cases, the diagnosis of C3GN was based on the presence of dominant/isolated IF deposition of C3. Interestingly, DNA analysis in our patient revealed similar CFB gene variant: c.1697A.C, p.Glu566Arg (an adenine to cytosine change at nucleotide 1697, leading to a substitution of glutamate by arginine at amino acid 566). Whether the renal injury and exaggerated alternative complement pathway with the accumulation of C3/complement degradation products are a result of an antibody–antigen immune-complex-triggering condition that activates the classic complement pathway and subsequently cross-talks with the alternative complement pathway or are secondary to c.1697A.C, p.Glu566Arg mutation in CFB is debatable, as the pathogenicity of this genetic defect is yet to be defined. Although p.Glu566Ala defect was reported in association with C3GN [10], Marinozzi et al. found this mutation to have partial effect on CFB function and represents a genetic pleomorphism in the setting of aHUS [12]. In our case, the pathogenicity of p.Glu566Arg heterozygous variant should be considered, especially with the presence of aHUS and progressive renal failure. Also, remarkable improvement in renal function, urine output, and serum C3 levels were seen only after three doses of eculizumab, which considerably denotes the pathogenicity of the genetic variant. However, it should also be noted that the same genetic variant in CFB was identified in the patient’s father, who demonstrated no evidence of laboratory or clinical abnormalities. Whether incomplete penetrance of the mutation and other genetic or environmental predisposing factors play a role in how the mutation is clinically manifested needs to be further explored [13, 14].
The MPGN pattern of glomerular injury and intense staining of C3 are features of C3 glomerulopathy. The presence of significant staining for IgG and light chains and intense staining for C1q—a component of the classic complement activation pathway commonly found in IC-MPGN—plays major role in the deposition and retention of antibody–antigen immune complexes in the GBM [15], consistent with the diagnosis of IC-MPGN in our patient. In addition, it should be kept in mind that in many glomerular diseases, including C3GN, small amounts of immunoglobulins and other immune reactants can be trapped in sclerotic glomerular tufts [16]. In our case, the strong IF intensity and well-defined subendothelial accumulation of IgG and C1q in the patent’s glomerular capillaries strongly favor MPGN and makes C3GN unlikely. The negative C4d immunocytochemistry and response to eculizumab and not to steroids and MMF therapy are intriguing facts and raise the possibility of alternative complement pathway dysregulation as an important factor in the pathogenesis of the renal injury, rather than immune-complex-mediated mechanism only.
Infection-related glomerulonephritis is most commonly manifested histologically as an acute diffuse proliferative exudative glomerulonephritis associated with glomerular immune complex deposits, which predominantly contain C3 and IgG and may cross-react with the alternate complement pathway [17]. Furthermore, patients with genetic defects in complement protein are susceptible to exacerbation of kidney disease during infections and C3-dominant acute proliferative glomerulonephritis following streptococcal infection, as perviously described in a patient with heterozygous CFHR-5 deficiency [18]. Whether our patient had an infection that triggered antigen–antibody complexes and eventually precipitated the complement mutation defects is not clear. Blood cultures were sterile, but blood samples were cultured 4 days after antibiotic administration, which may explain blood culture negativity. Interestingly, renal biopsy demonstrated severe exudative glomerulonephritis with exuberant cellular crescents, a pattern that has not been previously reported in association with genetic heterozygous variant of the CFB gene. The exudative nature of the glomerular injury with intense and abundant C3 deposition should raise the question of acute postinfectious glomerulonephritis (PIGN). However, negative ASO serology, nonresponse to antibiotic treatment, the presence of mesangial alteration, lobulation of glomerular tufts, double contouring of the GBM and subendothelial deposits by light microscopy, strong C1q IF positivity, and the presence of diffuse and abundant subendothelial immune-type dense deposits ultrastructurally made PIGN an unlikely histopathological diagnosis. Scattered discrete subendothelial immune-type dense deposits are seen in the early stage of classic PIGN and can be abundant in atypical PIGN, commonly associated with deep-seated abscess or shunt nephritis.
HUS is a triad of microangiopathy hemolytic anemia, thrombocytopenia, and acute renal failure [19]. Atypical (non-Shiga-like toxin) HUS occurs in sporadic cases secondary to a variety of precipitating conditions, such as viral and bacterial infections, autoimmune diseases, drugs, pregnancy, and allograft rejection, or it can be familial and associated with genetic abnormalities of the complement regulatory proteins, including C3, CFH, CFI, and CFB, with resultant overactivation of the alternative complement pathway on the endothelial surface and endothelial cell injury [2]. Renal biopsy often shows a thrombotic microangiopathy pattern of injury with no significant IF positivity of immunoglobulin or complements and absence of immune-dense deposits by ultrastructural study [20]. Various gain-of-function mutations that deregulate or hyperactivate C3 convertase in factor B (FB) ligand-binding sites were described in the association of aHUS [12]. We speculate that in our patient, progression of aHUS could be related to the pathogenicity of p.Glu566Arg heterozygous variant; however, functional assessment of this genetic variant was not performed.
In conclusion, we describe a rare heterozygous variant—p.Glu566Arg—in the CFB gene that was associated with IC-MPGN and aHUS, which responded clinically and biochemically to eculizumab. Screening of possible genetic defects in complement regulatory proteins may be considered in immune-complex-mediated glomerulonephritis triggering the classic complement pathway, particularly those associated with aHUS, as prompt diagnosis and appropriate management are vital to preserving renal function. Mutations in CFB pathogenicity of heterozygous variants are rarely reported. As functional significance is not available in our case, continuing eculizumab for life remains uncertain. Hence, reporting mutations in the CFB gene is necessary to further explore and expand our knowledge of the pathogenicity of different genetic variants.
References
Wong EK, Goodship TH, Kavanagh D (2013) Complement therapy in atypical hemolytic uraemic syndrome (aHUS). Mol Immunol 56(3):199–212
Kavanagh D, Goodship TH, Richards A (2013) Atypical hemolytic uremic syndrome. Semin Nephrol 33(6):508–530
Sartz L, Olin AI, Kristoffersson AC, Ståhl AL, Johansson ME, Westman K, Fremeaux-Bacchi V, Nilsson-Ekdahl K, Karpman D (2012) A novel C3 mutation causing increased formation of the C3 convertase in familial atypical hemolytic uremic syndrome. J Immunol 188(4):2030–2037
Tawadrous H, Maga T, Sharma J, Kupferman J, Smith RJ, Schoeneman M (2010) A novel mutation in the complement factor B gene (CFB) and atypical hemolytic uremic syndrome. Pediatr Nephrol 25(5):947–951
Leroy V, Fremeaux-Bacchi V, Peuchmaur M, Baudouin V, Deschênes G, Macher MA, Loirat C (2011) Membranoproliferative glomerulonephritis with C3NeF and genetic complement dysregulation. Pediatr Nephrol 26(3):419–424
Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, D’Agati VD (2014) Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int 85(2):450–456
Sethi S, Fervenza FC (2012) Membranoproliferative glomerulonephritis--a new look at an old entity. N Engl J Med 366(12):1119–1131
Volokhina E, Westra D, Xue X, Gros P, van de Kar N, van den Heuvel L (2012) Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol 27(9):1519–1524
Cayci FS, Cakar N, Hancer VS, Uncu N, Acar B, Gur G (2012) Eculizumab therapy in a child with hemolytic uremic syndrome and CFI mutation. Pediatr Nephrol 27(12):2327–2331
Imamura H, Konomoto T, Tanaka E, Hisano S, Yoshida Y, Fujimura Y, Miyata T, Nunoi H (2015) Familial C3 glomerulonephritis associated with mutations in the gene for complement factor B. Nephrol Dial Transplant 30(5):862–864
Sethi S, Smith RJ, Dillon JJ, Fervenza FC (2015) C3 glomerulonephritis associated with complement factor B mutation. Am J Kidney Dis 65(3):520–521
Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, Smith RJ, Noris M, Halbwachs-Mecarelli L, Donadelli R, Fremeaux-Bacchi V, Roumenina LT (2014) Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol 25(9):2053–2065
Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H (2013) Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 132(10):1077–1130
Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, López-Trascasa M, Sánchez-Corral P, Morgan BP, Rodríguez de Córdoba S (2007) Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 104(1):240–245
Bohnsack JF, Tenner AJ, Laurie GW, Kleinman HK, Martin GR, Brown EJ (1985) The C1q subunit of the first component of complement binds to laminin: a mechanism for the deposition and retention of immune complexes in basement membrane. Proc Natl Acad Sci U S A 82(11):3824–3828
Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, Doyle M, Fakhouri F, Fervenza FC, Fogo AB, Frémeaux-Bacchi V, Gale DP, Goicoechea de Jorge E, Griffin G, Harris CL, Holers VM, Johnson S, Lavin PJ, Medjeral-Thomas N, Paul Morgan B, Nast CC, Noel LH, Peters DK, Rodríguez de Córdoba S, Servais A, Sethi S, Song WC, Tamburini P, Thurman JM, Zavros M, Cook HT (2013) C3 glomerulopathy: consensus report. Kidney Int 84(6):1079–1089
Khalighi MA, Wang S, Henriksen KJ, Bock M, Keswani M, Meehan SM, Chang A (2016) Revisiting post-infectious glomerulonephritis in the emerging era of C3 glomerulopathy. Clin Kidney J 9(3):397–402
Vernon KA, Goicoechea de Jorge E, Hall AE, Fremeaux-Bacchi V, Aitman TJ, Cook HT, Hangartner R, Koziell A, Pickering MC (2012) Acute presentation and persistent glomerulonephritis following streptococcal infection in a patient with heterozygous complement factor H-related protein 5 deficiency. Am J Kidney Dis 60(1):121–125
Kavanagh D, Richards A, Atkinson J (2008) Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med 59:293–309
Sethi S, Fervenza FC (2014) Pathology of renal diseases associated with dysfunction of the alternative pathway of complement: C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost 40(4):416–421
Authors’ contributions
• Khalid Alfakeeh: Clinical management and follow-up, data collection and review of the manuscript.
• Mohammed Azar: Clinical management and follow-up, data collection and writing the manuscript.
• Majid Alfadhel: Comment on genetic analysis and review of the manuscript.
• Alsuayri Mansour Abdullah: Data collection.
• Nourah Aludah: Pathological interpretation, image capture, and manuscript review.
• Khaled O. Alsaad: Pathological interpretation, manuscript composition
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Written informed consent for the publication of this case report and any accompanying images has been obtained from the patient’s mother. A copy of the written consent is available for review by the editor.
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Alfakeeh, K., Azar, M., Alfadhel, M. et al. Rare genetic variant in the CFB gene presenting as atypical hemolytic uremic syndrome and immune complex diffuse membranoproliferative glomerulonephritis, with crescents, successfully treated with eculizumab. Pediatr Nephrol 32, 885–891 (2017). https://doi.org/10.1007/s00467-016-3577-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-016-3577-0