
Abstract
Background
We report the case of a patient with Shiga toxin (Stx)-associated hemolytic-uremic syndrome (HUS) (STEC-HUS) with a concomitant heterozygous mutation of the gene coding for complement Factor H (CFH).
Case diagnosis/treatment
An 18-month-old patient presented with hemolytic anemia and thrombotic microangiopathy in the context of acute gastroenteritis. While the patient did not show kidney or other organ failure, he had persistent hemolysis and complement 3 activation (low C3), leading to the decision to commence immunotherapy with eculizumab (Soliris®) together with transient antibiotic coverage and meningococcal vaccination. Patient outcome was favorable. Diagnostic work-up identified Escherichia coli-associated Type 2 Shiga toxin. Complement analysis showed a heterozygous mutation of the CFH gene (c.2103 G>A, p. Trp701X) resulting in a quantitative CFH defect.
Conclusions
We report a case of STEC-HUS with a quantitative CFH defect caused by a mutation of the CFH gene. To the best of our knowledge, very few cases of STEC-HUS with complement gene mutation have been reported, but none to date with a CFH mutation. We therefore suggest that complement analyses be performed in patients diagnosed with STEC-HUS in association with low C3 levels, especially in patients presenting with severe or unexpected clinical symptoms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Hemolytic uremic syndrome (HUS) is a non-immune disease characterized by hemolytic anemia, low platelet count and renal impairment [1]. There are two types of HUS. The first is secondary to Shiga-like toxin (Stx)-producing Escherichia coli (STEC). While bacteria-induced bloody diarrhea develops in the majority of patients with STEC-HUS, the prevalence of cases without diarrhea is reported to be 25 % [1]. STEC-HUS is most frequent in children, with an annual incidence of 6.1 cases per 100,000 children under 5 years of age [1, 2]. The second type of HUS, representing 10 % of all cases, is called atypical hemolytic uremic syndrome (aHUS) [1, 2]. aHUS is a heterogeneous disease which is associated with an uncontrolled activation of the complement alternative pathway (AP) in more than half of cases [2, 3]. This complex complement-mediated disease induces thrombotic microangiopathy with possible lesions in kidney, brain, heart, lung, gastro-intestinal tract and pancreas [2]. aHUS can be sporadic or familial [4], and there is a link between aHUS and genetic abnormalities in complement AP regulatory genes, including complement Factor H (CFH), Factor I (CFI), C3, membrane cofactor protein (MCP/CD46) and Factor B (CFB) [4, 5]. The prognosis of patients with aHUS is poor compared to that of patients with STEC-HUS, with more progression to end-stage renal disease (ESRD) (>50 %) and higher mortality (6.8 % during the first year in children in a French cohort) among the former [2, 5].
Despite recent in vivo [6, 7] and in vitro [8, 9] evidence for complement involvement in STEC-HUS, complement work-up is currently not felt to be necessary in patients with STEC-HUS. Here we report a child with STEC-HUS and a heterozygous CFH mutation resulting in low systemic CFH levels and complement AP overactivation.
Case report
A previously healthy 18-month-old boy with a non-contributory (personal and family) medical history was hospitalized for jaundice, pallor, vomiting, non-bloody diarrhea and weight loss. The laboratory work-up identified hemolytic anemia with a hemoglobin count of of 4.5 g/dL, hematocrit of 12.8 %, lactate dehydrogenase (LDH) level of 2448 U/L, schizocytes and a very low haptoglobin level. His platelet count was 20,000/μL, and he presented with acute renal failure (serum creatinine 99 μmol/L, urea 14.4 mmol/L). Although renal function was impaired, diuresis was preserved. Systolic and diastolic blood pressures were on the 99th percentile [10]. There were no extra-renal manifestations, such as neurological complications or cardiac failure. The background of acute gastroenteritis led to the suspicion of HUS. Despite the low platelet level, a diagnosis of thrombotic thrombocytopenic purpura was not supported, especially in the absence of neurological symptoms [11]. Two red cell transfusions were given, and azithromycin treatment was commenced in accordance with our local protocol [12].
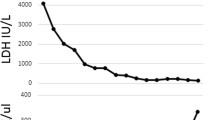
Complement component 3 (C3) levels were low (0.52 g/L; normal range 0.69–1.32 g/L) with normal C4 and total complement levels. Give the patient’s persistent hemolysis, decreased C3 level and persistent acute kidney injury, aHUS was suspected despite the presence of diarrhea. To avoid dialysis, two doses of eculizumab (600 and 300 mg on consecutive days) were given starting on the third day after diagnosis. This treatment was performed under antibiotic and vaccination coverage. Treatment response was favorable: platelet count was normalized at day 9 of the diagnosis, diuresis and blood pressure stayed normal, the patient did not require dialysis and renal function had normalized by the end of day 10 (Fig. 1). One month after HUS diagnosis, C3 level was normalized (Fig. 1) and hemoglobin was 11.5 g/dL without schizocytes and with normal haptoglobin level. Blood pressure was below the 95th percentile [10].

Biological parameters of the patient during the course of the disease. LDH Lactate dehydrogenase, C3 complement component 3
Of note, stool analysis was positive for STEC, and Stx2 was identified by PCR analysis, confirming the diagnosis of STEC-HUS.
Given the low C3 levels, a complete genetic analysis of complement AP activators/regulators was performed in a reference center. CFH serum levels [determined using enzyme-linked immunosorbent assays (ELISA)] were low on two distinct samples (40 and 57 %, respectively; normal range 65–140 %), both measured at diagnosis and before the start of eculizumab treatment. Genetic analysis revealed a heterozygous CFH gene mutation (c.2103 G>A, p. Trp701X). It is known that this abnormality leads to CFH deficiency as the mutated gene is unable to encode circulating protein due to a stop codon. Accordingly, in our patient this heterozygous mutation was associated with low C3 levels and low CFH serum levels. Multiplex ligation-dependent probe amplification screening of the CFH-related (CFHR) locus, including screening for the recently published hybrid CFH/CFHR1 gene, was negative [13]. However, the homozygous haplotype CFH tgtgt, defined by a single nucleotide polymorphism database as being at increased prevalence in patients with aHUS than in healthy controls, was isolated [2, 14]. Further complement gene analysis (including MCP/CD46, CFI, CFB) was negative. There were no anti-CFH autoantibodies. Factor I level and ADAMTS-13 activity were normal.
The follow-up at 18 months after initial presentation showed a normal renal function, without hemolysis (hemoglobin 12.7 g/dL, haptoglobin 0.52 g/L, LDH 294 UI/mL). C3 level was near the lower limit of normal (0.67 g/L; normal range 0.69–1.32 g/L). The patient is still being followed up at our center.
We suggested to the parents that they also be screened for this mutation, but they refused. They have no medical history of renal disease or hemolysis.
Discussion and conclusion
We report a patient with STEC-HUS, without severe organ failure and a good outcome. Complement analysis identified a heterozygous CFH mutation (c.2103 G>A, p. Trp701X) with low CFH serum levels.
CFH is the most important regulatory protein in the complement AP [4]. It is one of the CFI cofactors, each of which contributes to inhibiting the formation of the AP C3 convertase (C3bBpP) [15]. More than 100 CFH mutations have been reported to date, of which the majority are heterozygous [4, 5, 16]. There are two types of CFH mutations. Type II CFH mutations differ from Type I mutations which are associated with a decreased CFH serum level [5, 15]. Our patient’s CFH gene mutation is a Type I mutation, but one that is not included in any database. This novel mutation leads to heterozygous CFH deficiency, which has been described to be associated with C3 consumption by the complement AP [17].
aHUS occurs more frequently in adults [1], but aHUS with CFH mutations is more common in children [5, 18]. aHUS has a poor prognosis, with death or progression to ESRD in 60 % of cases within the first year of life [5, 16]. Of note, creatinine levels during the first aHUS episode are predictive of prognosis [16].
Some triggering events are well known for aHUS, such as infections (upper respiratory tract infection, fever, diarrhea), malignancies, drugs, pregnancy and underlying medical infection [1, 4]. Complement mutation is a risk factor for aHUS, but it is not the direct cause [4, 16]. Identification of an underlying complement mutation is helpful in the determination of the prognosis, treatment duration and management of recurrence [5].
With the advent of eculizumab, aHUS treatment has recently undergone a significant paradigm shift. Eculizumab is a monoclonal humanized anti-C5 antibody that prevents the activation of C5 and, consequently, of the terminal complement pathway with the formation of the C5b9 lytic membrane attack complex [4, 15]. Eculizumab increases the risk of Neisseria meningitis infection, and specific vaccination in combination with transient antibiotic coverage is therefore mandatory at the time of treatment initiation [15]. Eculizumab has been successfully used in pediatric and adult patients with aHUS [19], as well as in children with STEC HUS with severe extrarenal, mainly central nervous system, manifestations who were refractory to conservative therapy and plasma treatment [20, 21]. This treatment benefit is in keeping with recent in vivo observations of evidence for activation of the complement AP during the acute phase of STEC-HUS [6, 7] and in vitro findings indicating that Stx is capable of directly activating the complement system [8, 9].
Indeed, complement seems to be involved in STEC-HUS in various ways [9, 22–28]. Morigi et al. demonstrated both in vivo and in vitro that complement activation via P-selectin, whose expression on the endothelial cell surface is induced by Stx, is a key mechanism of C3a-dependent microvascular thrombosis in STEC-HUS [22]. Along the same lines, Locatelli et al. suggested that generation of C3a in STEC-HUS promotes an integrin-linked kinase signaling that leads to podocyte dysfunction and loss [23]. Other explanations include a role for EspP, a serine protease of E. coli, which could induce complement downregulation [25]. Ehrlenbach et al. showed that Stx2 reduces CD59 expression on the surface of human tubular epithelial and glomerular endothelial cells [24]. CD59 could play a role in protecting glomeruli from complement attack. Some authors have shown that there is probably complement activation in the circulation during STEC-HUS, firstly on red blood cells, which could play a role in STEC-HUS hemolysis, and secondly on blood cells and blood cell-derived microparticles [26, 27].
Previous studies have demonstrated that Stx activates the complement AP, although the exact physiopathology remains to be determined. It has also been demonstrated that Stx can inhibit complement’s most potent fluid phase regulator, CFH [8, 9, 28]. Stx seems to be an important trigger of aHUS in a patient who has a CFH defect [15]. On the other hand, Paixao-Cavalcante et al. showed that Stx2 leads to tubular injury in CFH-deficient mice, but also in wild mice [29]. In their study, renal histology of the mice did not show thrombotic microangiopathy, probably because Stx globotriaosylceramide (Gb3) receptors in mouse kidneys are located in the tubular and collecting duct epithelial cells, and not in glomeruli. Therefore, these authors were not able to confirm their hypothesis that CFH deficiency increases host susceptibility to STEC-HUS.
We report on the successful use of early treatment with eculizumab in a patient with STEC-HUS, but we cannot be sure that our patient would not have spontaneously recovered. Treatment escalation was motivated by persistent hemolysis and a low C3 serum level, as well as to avoid dialysis. Of note, our patient was subsequently identified as having an underlying CFH mutation (c.2103 G>A, p. Trp701X). Thus, we present one of the first cases of STEC-HUS complicated by a defect in the regulation of the complement AP. Our findings have significant implications: (1) even though likely rare, the possibility of an aggravating underlying complement mutation should be considered even in patients with STEC-HUS, and (2) we provide further rationale for the use of complement-targeting treatment in STEC-HUS. While it is premature to claim eculizumab as a standard treatment in STEC-HUS, even in cases of low C3 serum levels, there may be a rationale for a randomized controlled trial of its use in such patients.
aHUS patients can have decreased C3 levels with normal C4 levels [2]. Low C3 levels indicate an activation of the complement AP [2, 5], and they are more frequent in patients with CFH or C3 mutations than in patient without mutations [18]. However, normal C3 levels do not exclude complement dysfunction or predict a favorable outcome [1, 16].
Based on our experience, we propose performing complement analyses in STEC-HUS patients with low C3 levels, even mildly low levels, as low C3 levels may be induced by C3 consumption, such as secondary to CFH deficiency [30]. To support this proposal, a study with systematic complement analysis in current and former STEC-HUS patients would be interesting.
Our patient had STEC and a CFH mutation. To our knowledge, there are very few reports of patients with STEC-HUS and a complement gene mutation. Fremeaux-Bacchi et al. studied 214 aHUS patients who had complement abnormalities [5]. It has also been reported that three patients who had STEC-HUS at the first episode had subsequent relapses, suggesting the diagnosis of aHUS. Alberti et al. described two adult patients with heterozygous complement mutations (MCP/CD46 and CFI) who initially were diagnosed with STEC-HUS, but who had HUS recurrence in their transplanted kidney [31]. The MCP/CD46 and CFI mutations were identified only after HUS recurrence. The heterozygous MCP/CD46 mutation was identified in the patient’s mother who had donated the kidney, explaining the recurrence post-transplant and leading the authors to propose pre-transplant genetic counseling in patients who develop ESRD following STEC-HUS.
The cases reported in this context bring up an interesting discussion of the applicable terminology: as complement abnormalities can be considered more as a risk factor than as the cause of HUS, these cases would by the current definition be termed STEC and not aHUS. On the other hand, the underlying complement mutations will possibly aggravate disease precipitation and render the patients susceptible to treatment with complement-targeting treatments, such as eculizumab. Therefore, Loirat et al. stated the hypothesis that the classical classification of diarrhea or non-diarrhea HUS could be misleading because post-diarrheal onset does not exclude genetic aHUS [4]. Our case and those reported in the literature consolidate this hypothesis [5, 16, 31].
In conclusion, we interpret our patient with a CFH mutation as a case of aHUS triggered by a STEC infection. The classification of a patient classification as STEC-HUS or aHUS should be done carefully. Complement analyses seem to be necessary, particularly when CFH or C3 levels are low, even if STEC is isolated in the stool. Insight gained via these studies might become useful to inform the treatment choice, especially in cases of disease recurrence.
References
Noris M, Remuzzi G (2005) Hemolytic uremic syndrome. J Am Soc Nephrol 16:1035–1050
Noris M, Remuzzi G (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361:1676–1687
Waters AM, Licht C (2011) aHUS caused by complement dysregulation: new therapies on the horizon. Pediatr Nephrol 26:41–57
Loirat C, Noris M, Fremeaux-Bacchi V (2008) Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol 23:1957–1972
Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschênes G, Lebranchu Y, Zuber J, Loirat C (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554–562
Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Lesser M, Kline M, Hoffman C, Christen E, Trachtman H (2009) Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 4:1920–1924
Ferraris JR, Ferraris V, Acquier AB, Sorroche PB, Saez MS, Ginaca A, Mendez CF (2015) Activation of the alternative pathway of complement during the acute phase of typical hemolytic uremic syndrome. Clin Exp Immunol 181:118–125
Orth D, Wurzner R (2010) Complement in typical hemolytic uremic syndrome. Semin Thromb Hemost 36:620–624
Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Würzner R (2009) Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 182:6394–6400
National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents (2004) The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 114[2 Suppl 4th Report]:555–576
Trachtman H (2013) HUS and TTP in children. Pediatr Clin N Am 60:1513–1526
Borgatta B, Kmet-Lunacek N, Rello J (2012) E. coli O104:H4 outbreak and haemolytic-uraemic syndrome. Med Intensiva 36:576–583
Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, Diaz-Torres ML, Sampson A, Mead P, Webb M, Pirson Y, Jackson MS, Huguess A, Woods KM, Goodship JA, Goodship THJ (2006) Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med 3:e431
Malina M, Roumenina LT, Seeman T, Le Quintrec M, Dragon-Durey MA, Schaefer F, Fremeaux-Bacchi V (2012) Genetics of hemolytic uremic syndromes. Presse Med 41:e105–114
Roumenina LT, Loirat C, Dragon-Durey MA, Halbwachs-Mecarelli L, Sautes-Fridman C, Fremeaux-Bacchi V (2011) Alternative complement pathway assessment in patients with atypical HUS. J Immunol Methods 365:8–26
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschênes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C, Loirat C (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 18:2392–2400
Dragon-Durey MA, Fremeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschênes G, Coppo P, Herman Fridman W, Weiss L (2004) Heterozygous and homozygous Factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis : report and genetic analysis of 16 cases. J Am Soc Nephrol 15:787–795
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, Van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5:1844–1859
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham CC, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fougue D, Furman RR, Gaber O, Herthelius M, Houmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nürnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368:2169–2181
Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx MJ, Le Deist F, Niaudet P, Schaefer F (2011) Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med 364:2561–2563
Braune SA, Wichmann D, von Heinz MC, Nierhaus A, Becker H, Meyer TN, Meyer GP, Müller-Schulz M, Fricke J, De Weerth A, Hoepker WW, Fiehler J, Magnus T, Gerloff C, Panzer U, Stahl RAK, Wegsheider K, Kluge S (2013) Clinical features of critically ill patients with Shiga toxin-induced hemolytic uremic syndrome. Crit Care Med 41:1702–1710
Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C (2011) Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 187:172–180
Locatelli M, Buelli S, Pezzotta A, Corna D, Perico L, Tomasoni S, Rottoli D, Rizzo P, Conti D, Thurman JM, Remuzzi G, Zoja C, Morigi M (2014) Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J Am Soc Nephrol 25:1786–1798
Ehrlenbach S, Rosales A, Posch W, Wilflingseder D, Hermann M, Brockmeyer J, Karch H, Satchell SC, Würzner R, Orth-Höller D (2013) Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect Immun 81:2678–2685
Orth D, Ehrlenbach S, Brockmeyer J, Khan AB, Huber G, Karch H, Sarg B, Lindner H, Würzner R (2010) EspP, a serine protease of enterohemorrhagic Escherichia coli, impairs complement activation by cleaving complement factors C3/C3b and C5. Infect Immun 78:4294–4301
Arvidsson I, Stahl AL, Hedstrom MM, Kristoffersson AC, Rylander C, Westman JS, Storry JR, Olsson ML, Karpman D (2015) Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 194:2309–2318
Stahl AL, Sartz L, Karpman D (2011) Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 117:5503–5513
Poolpol K, Orth-Holler D, Speth C, Zipfel PF, Skerka C, de Cordoba SR, Brockmeyer J, Bielaszewska M, Würzner R (2014) Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol 58:77–84
Paixao-Cavalcante DBM, Cook HT, Pickering MC (2008) Shiga toxin-2 results in renal tubular injury but not thrombotic microangiopathy in heterozygous factor H-deficient mice. Clin Exp Immunol 155:339–347
Noris M, Ruggenenti P, Perna A, Orisio S, Caprioli J, Skerka C, Vasile B, Zipfel PF, Remuzzi G (1999) Hypocomplementemia discloses genetic predisposition to hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: role of factor H abnormalities. Italian Registry of Familial and Recurrent Hemolytic Uremic Syndrome/Thrombotic Thrombocytopenic Purpura. J Am Soc Nephrol 10:281–293
Alberti M, Valoti E, Piras R, Bresin E, Galbusera M, Tripodo C, Thaiss F, Remuzzi G, Noris M (2013) Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant 13:2201–2206
Conflict of interest
The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Caillaud, C., Zaloszyc, A., Licht, C. et al. CFH gene mutation in a case of Shiga toxin-associated hemolytic uremic syndrome (STEC-HUS). Pediatr Nephrol 31, 157–161 (2016). https://doi.org/10.1007/s00467-015-3207-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-015-3207-2